*Bartosz Chyżyński, Barbara Sikorska-Fic, Edyta Niewiadomska, Michał Matysiak
Zespół rozsianego wykrzepiania wewnątrznaczyniowego w ostrych białaczkach – patofizjologia, obraz kliniczny, diagnostyka oraz leczenie
Disseminated intravascular coagulation in acute leukemia – pathophysiology, clinical picture, diagnostics and treatment
Katedra i Klinika Pediatrii, Hematologii i Onkologii, Warszawski Uniwersytet Medyczny
Kierownik Kliniki: prof. dr hab. n. med. Michał Matysiak
Summary
Disseminated intravascular coagulation syndrome (DIC) is a syndrome characterized by generalized intravascular activation of the coagulation system. This syndrome is not an isolated clinical syndrome, but is always secondary to other diseases. It is particularly often observed in neoplastic diseases, and in particular in haematopoietic malignancies such as acute leukemia. The risk of DIC depends on the type and subtype of acute leukemia. DIC is most often observed in the course of acute myeloid leukaemia with particular emphasis on the subtype M3 and M5 according to FAB. The basic treatment for DIC remains the treatment of the underlying disease and substitution treatment. Depending on the disease underlying the development of DIC, the substitution procedure may differ significantly. Knowledge of the pathomechanism of disorders occurring in DIC is the key to early diagnosis and rapid implementation of treatment.

Wstęp
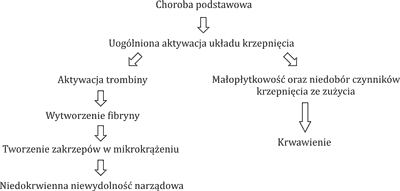
Zespół rozsianego wykrzepiania wewnątrznaczyniowego (ang. disseminated intravascular coagulation – DIC) jest stanem charakteryzującym się uogólnioną wewnątrznaczyniową aktywacją układu krzepnięcia prowadzącą do wytworzenia dużej ilości fibryny. Konsekwencją tego procesu jest formowanie się mikrozakrzepów, które upośledzają przepływ krwi w mikrokrążeniu, doprowadzając do niewydolności narządowej. Dodatkowo, w wyniku masywnego i postępującego wykrzepiania wewnątrznaczyniowego dochodzi do stopniowego zużycia płytek krwi oraz osoczowych czynników krzepnięcia, co może doprowadzić do objawów skazy krwotocznej pod postacią krwawień. DIC nie jest izolowanym zespołem klinicznym, lecz zawsze towarzyszy innym jednostkom chorobowym (tab. 1) (1, 2). Może on przebiegać w sposób jawny klinicznie, a w sposób szczególnie nasilony np. u krytycznie chorych pacjentów. W wielu przypadkach zaburzenie to nie daje jednak objawów ani też odchyleń w standardowo wykonywanych badaniach laboratoryjnych. DIC może być wtedy wykryte jedynie za pomocą czułych i specyficznych testów badających układ krzepnięcia. Znajomość patomechanizmu zaburzeń charakterystycznych dla tego zespołu jest kluczem do wdrożenia szybkiej diagnostyki i skutecznego leczenia pacjentów z grup ryzyka wystąpienia DIC (tab. 1).
Tab. 1. Stany chorobowe przebiegające z DIC (na podstawie (3))
| Sepsa, ciężkie infekcje |
| Uraz, oparzenie, złamanie |
Nowotwory
– Guzy lite
– Ostre białaczki |
Schorzenia położnicze
– Zatory wodami płodowymi
– Oddzielenie łożyska
– Zespół HELLP (hemoliza, podwyższona aktywność enzymów wątrobowych, małopłytkowość) |
Nieprawidłowości naczyniowe
– Zespół Kasbach-Meritt
– Malformacje naczyniowe
– Tętniak aorty |
| Ciężkie reakcje alergiczne |
| Ciężkie reakcje immunologiczne, np. reakcja poprzetoczeniowa |
Patofizjologia
Podłożem zaburzeń obserwowanych w DIC jest nadmierne pobudzenie trombogenezy z jednoczesnym upośledzeniem funkcji układu fibrynolizy.
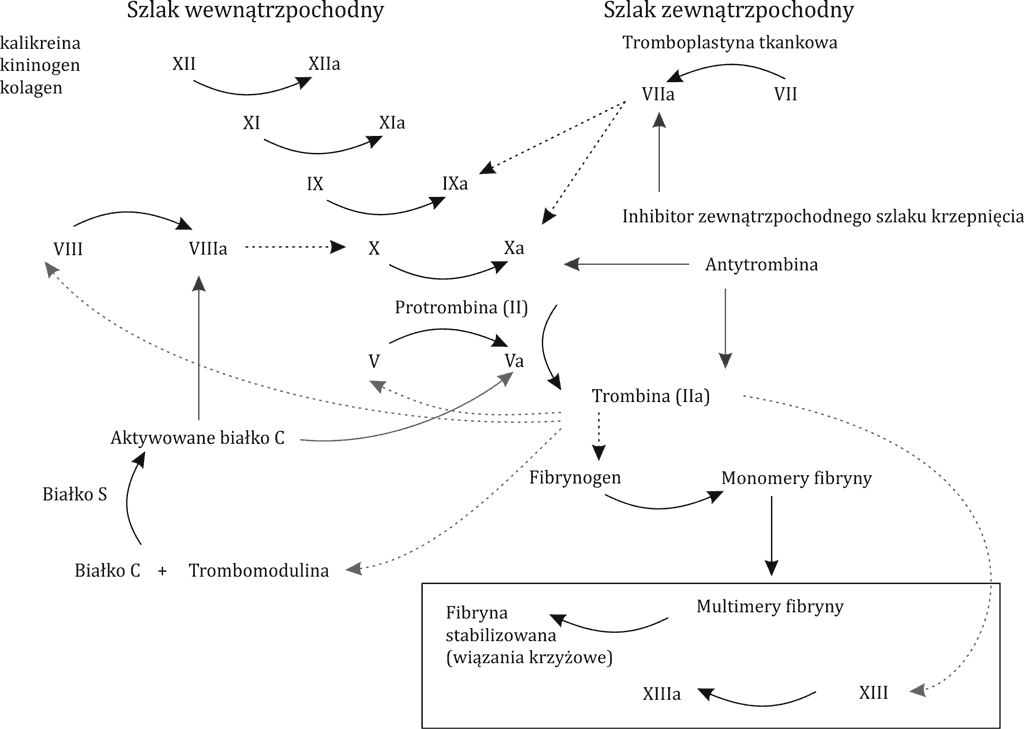
W przebiegu DIC kluczowym mechanizmem obserwowanych zaburzeń okazuje się aktywacja układu krzepnięcia na drodze zewnętrznej. Pod wpływem takich czynników, jak: interleukiny, czynnik martwicy nowotworów (TNF), endotoksyny, prokoagulant rakowy (CP), dochodzi do wzmożonej aktywacji trombiny w szlaku zależnym od czynnika tkankowego (TF), co prowadzi do zwiększonej produkcji fibryny. Droga wewnętrzna aktywacji układu krzepnięcia wydaje się nie odgrywać istotnej roli w aktywacji trombogenezy w DIC (4, 5). W DIC stwierdza się zmniejszenie aktywności endogennych inhibitorów krzepnięcia. Często obserwuje się obniżenie stężenia antytrombiny III (AT III), które wynika zarówno ze zużycia w przebiegu globalnej aktywacji trombiny, jak również z jej rozkładu przez elastazy i proteazy wytwarzane przez neutrofile, a także z zaburzonej produkcji inhibitora trombiny (2, 6). Innym białkiem, którego niedobór stwierdzany jest w procesie uogólnionej aktywacji krzepnięcia, jest białko C. Jego zmniejszona produkcja wynika z upośledzonej ekspresji trombomoduliny na komórkach śródbłonka naczyniowego pod wpływem cytokin prozapalnych, co dodatkowo potęguje stan prozakrzepowy (7). Kolejną przyczyną DIC jest zahamowanie funkcji układu fibrynolizy. Obserwuje się znaczną aktywację procesów hamujących fibrynolizę w wyniku zwiększonej produkcji inhibitora aktywatora plazminogenu 1 (PAI-1) (2, 8). W przebiegu DIC istnieje dwukierunkowa zależność między cytokinami prozapalnymi a elementami układu krzepnięcia. Z jednej strony interleukiny TNF-α prowadzą do aktywacji układu krzepnięcia w mechanizmie opisanym powyżej, a jednocześnie procesy krzepnięcia aktywują komórki śródbłonka naczyniowego, pobudzając je do produkcji cytokin prozapalnych, prowadząc do wytworzenia samonapędzającego się błędnego koła (2, 5). Uogólniona aktywacja układu krzepnięcia prowadzi do stopniowego zużycia płytek krwi, osoczowych czynników krzepnięcia, inhibitorów krzepnięcia, co w konsekwencji może powodować rozwój powikłań krwotocznych, w tym ciężkie zagrażające życiu krwawienia, np. do ośrodkowego układu nerwowego (OUN). Przyczyną DIC w ostrej białaczce szpikowej (AML) jest uwalnianie z blastów substancji prokoagulacyjnych, takich jak: czynnik tkankowy, prokoagulant rakowy, oraz cytokin, np. interleukiny 1 (IL-1), TNF-α, czynnika przepuszczalności naczyń (VPF). Cytokiny mogą stymulować monocyty do produkcji TF i modulują właściwości hemostatyczne komórek śródbłonka, przyczyniając się dodatkowo do aktywacji układu krzepnięcia. Odpowiedzią na aktywację kaskady krzepnięcia jest wtórna fibrynoliza. Nieco inny mechanizm zaburzeń krzepnięcia stwierdzany jest w przebiegu ostrej białaczki promielocytowej (APL), gdzie koagulopatia wynika z dwóch współistniejących procesów: aktywacji układu krzepnięcia indukowanego przez czynnik tkankowy i prokoagulant rakowy oraz z nadmiernej aktywacji pierwotnej fibrynolizy. Ta ostatnia uważana jest za główną przyczynę obserwowanych zaburzeń krzepnięcia w momencie rozpoznania APL i jednocześnie krwawień, będących główną przyczyną wczesnej śmiertelności w przebiegu tego podtypu ostrej białaczki szpikowej. Powodem aktywacji układu fibrynolitycznego jest nadmierna ekspresja aneksyny II na powierzchni zmienionych nowotworowo promielocytów. Aneksyna II jest receptorem dla plazminogenu, jak również dla jego aktywatora tkankowego (tPA) (9, 10). Wiążąc te cząsteczki, jednocześnie potęguje około 60-krotnie produkcję plazminy, co prowadzi do nasilonej degradacji fibryny (11, 12). W ten sposób dochodzi do tzw. spadku stężenia fibrynogenu w osoczu i wynikających z tego częstych powikłań krwotocznych (ryc. 1).
Ryc. 1. Patofizjologia DIC (na podstawie (3))
Obraz kliniczny
Zespół DIC może przebiegać w postaci ostrej (jawny zespół DIC) lub przewlekłej (utajony zespół DIC). Obraz zaburzeń zależy od choroby podstawowej, w przebiegu której zespół DIC jest obserwowany oraz od wydolności mechanizmów kompensacyjnych, tj. wzmożonej syntezy czynników krzepnięcia w wątrobie, nasilonej trombopoezy w szpiku, jak również zwiększonego klirensu produktów degradacji fibryny (1). W postaci jawnej DIC obserwowane są często krwawienia z miejsc wkłuć obwodowych, nasilone krwawienia ze śluzówek jamy ustnej, przewodu pokarmowego, dróg oddechowych oraz w skrajnych przypadkach do OUN. Jednocześnie rozsiane zmiany zakrzepowe prowadzić mogą do niewydolności wielonarządowej (niewydolność wątroby, niewydolność oddechowa, niewydolność nerek, zaburzenia funkcji OUN). W postaci utajonej mamy do czynienia z miernie nasilonymi objawami skazy krwotocznej w postaci nawracających krwawień z nosa i/lub śluzówek jamy ustnej oraz tendencją do łatwego powstawania wylewów podskórnych. Ta postać DIC przebiega niejednokrotnie całkowicie bezobjawowo, a jedynym dowodem na trwające wykrzepianie wewnątrznaczyniowe są obserwowane zaburzenia w wynikach badań laboratoryjnych.
Rozpoznanie
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Windyga J: Patofizjologia, rozpoznawanie i leczenie rozsianego krzepnięcia wewnątrznaczyniowego. Hematologia 2011; 2(4): 326-331.
2. Levi M: Current understanding of disseminated intravascular coagulation. Br J Haematol 2004; 124(5): 567-576.
3. Levi M, Toh CH, Thachil J, Watson HG: Guidelines for the diagnosis and management of disseminated intravascular coagulation. Br J Haematol 2009; 145(1): 24-33.
4. van der Poll T, Buller HR, ten Cate H et al.: Activation of coagulation after administration of tumor necrosis factor to normal subjects. N Engl J Med 1990; 322(23): 1622-1627.
5. van der Poll T, de Jonge E, Levi M: Regulatory role of cytokines in disseminated intravascular coagulation. Semin Thromb Hemost 2001; 27(6): 639-651.
6. Mesters RM, Mannucci PM, Coppola R et al.: Factor VIIa and antithrombin III activity during severe sepsis and septic shock in neutropenic patients. Blood 1996; 88(3): 881-886.
7. Conway EM, Rosenberg RD: Tumor necrosis factor suppresses transcription of the thrombomodulin gene in endothelial cells. Mol Cell Biol 1988; 8(12): 5588-5592.
8. Biemond BJ, Levi M, Ten Cate H et al.: Plasminogen activator and plasminogen activator inhibitor I release during experimental endotoxaemia in chimpanzees: effect of interventions in the cytokine and coagulation cascades. Clin Sci (Lond) 1995; 88(5): 587-594.
9. Liu Y, Wang Z, Jiang M et al.: The expression of annexin II and its role in the fibrinolytic activity in acute promyelocytic leukemia. Leuk Res 2011; 35(7): 879-884.
10. Cesarman GM, Guevara CA, Hajjar KA: An endothelial cell receptor for plasminogen/tissue plasminogen activator (t-PA). II. Annexin II-mediated enhancement of t-PA-dependent plasminogen activation. J Biol Chem 1994; 269(33): 21198-21203.
11. Breen KA, Grimwade D, Hunt BJ: The pathogenesis and management of the coagulopathy of acute promyelocytic leukaemia. Br J Haematol 2012; 156(1): 24-36.
12. Ikezoe T: Pathogenesis of disseminated intravascular coagulation in patients with acute promyelocytic leukemia, and its treatment using recombinant human soluble thrombomodulin. Int J Hematol 2014; 100(1): 27-37.
13. Lee HJ, Park HJ, Kim HW, Park SG: Comparison of laboratory characteristics between acute promyelocytic leukemia and other subtypes of acute myeloid leukemia with disseminated intravascular coagulation. Blood Res 2013; 48(4): 250-253.
14. Sikorska-Fic B, Chyżyński B, Matysiak M: Two children with different clinical course for disseminated intravascular coagulation (DIC) on acute monoblastic leukemia (AML-M5) diagnosis. Pediatr Pol 2017; 92(3): 325-329.
15. Chojnowski K: Hemostasis disorders in acute leukemias. Acta Haematol Pol 2002; 33(2): 139-151.
16. Mitrovic M, Suvajdzic N, Bogdanovic A et al.: International Society of Thrombosis and Hemostasis Scoring System for disseminated intravascular coagulation > 6: a new predictor of hemorrhagic early death in acute promyelocytic leukemia. Med Oncol 2013; 30(1): 478.
17. Uchiumi H, Matsushima T, Yamane A et al.: Prevalence and clinical characteristics of acute myeloid leukemia associated with disseminated intravascular coagulation. Int J Hematol 2007; 86(2): 137-142.
18. Libourel EJ, Klerk CP, van Norden Y et al.: Disseminated intravascular coagulation at diagnosis is a strong predictor for both arterial and venous thrombosis in newly diagnosed acute myeloid leukemia. Blood 2016; 128(14): 1854-1861.
19. Higuchi T, Toyama D, Hirota Y et al.: Disseminated intravascular coagulation complicating acute lymphoblastic leukemia: a study of childhood and adult cases. Leuk Lymphoma 2005; 46(8): 1169-1176.
20. Di Bona E, Avvisati G, Castaman G et al.: Early haemorrhagic morbidity and mortality during remission induction with or without all-trans retinoic acid in acute promyelocytic leukaemia. Br J Haematol 2000; 108(4): 689-695.
21. Fenaux P, Castaigne S, Dombret H et al.: All-transretinoic acid followed by intensive chemotherapy gives a high complete remission rate and may prolong remissions in newly diagnosed acute promyelocytic leukemia: a pilot study on 26 cases. Blood 1992; 80(9): 2176-2181.
22. Altman JK, Rademaker A, Cull E et al.: Administration of ATRA to newly diagnosed patients with acute promyelocytic leukemia is delayed contributing to early hemorrhagic death. Leuk Res 2013; 37(9): 1004-1009.
23. Breccia M, Lo Coco F: Thrombo-hemorrhagic deaths in acute promyelocytic leukemia. Thromb Res 2014; 133 (suppl. 2): S112-116.
24. Stein E, McMahon B, Kwaan H et al.: The coagulopathy of acute promyelocytic leukaemia revisited. Best Pract Res Clin Haematol 2009; 22(1): 153-163.
25. Sanz MA, Grimwade D, Tallman MS et al.: Management of acute promyelocytic leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood 2009; 113(9): 1875-1891.
26. Rodeghiero F, Avvisati G, Castaman G et al.: Early deaths and anti-hemorrhagic treatments in acute promyelocytic leukemia. A GIMEMA retrospective study in 268 consecutive patients. Blood 1990; 75(11): 2112-2117.
27. Alimoghaddam K, Ghavamzadeh A, Jahani M: Use of Novoseven for arsenic trioxide-induced bleeding in PML. Am J Hematol 2006; 81(9): 720.
28. Yilmaz D, Karapinar B, Balkan C et al.: Single-center experience: use of recombinant factor VIIa for acute life-threatening bleeding in children without congenital hemorrhagic disorder. Pediatr Hematol Oncol 2008; 25(4): 301-311.
29. Takezako N, Sekiguchi N, Nagata A et al.: Recombinant human thrombomodulin in the treatment of acute myeloid leukemia patients complicated by disseminated intravascular coagulation: retrospective analysis of outcomes between patients treated with heparin and recombinant human thrombomodulin therapy. Thromb Res 2015; 136(1): 20-23.
30. Ikezoe T, Yang J, Nishioka C et al.: Thrombomodulin enhances the antifibrinolytic and antileukemic effects of all-trans retinoic acid in acute promyelocytic leukemia cells. Exp Hematol 2012; 40(6): 457-465.
31. Avvisati G, ten Cate JW, Buller HR, Mandelli F: Tranexamic acid for control of haemorrhage in acute promyelocytic leukaemia. Lancet 1989; 2(8655): 122-124.
32. Hashimoto S, Koike T, Tatewaki W et al.: Fatal thromboembolism in acute promyelocytic leukemia during all-trans retinoic acid therapy combined with antifibrinolytic therapy for prophylaxis of hemorrhage. Leukemia 1994; 8(7): 1113-1115.