Janusz Popowski
Choroby prionowe. Realne zagrożenie czy nieuzasadniona psychoza
Prion diseases. The real threat or groundless psychosis
Samodzielna Pracownia Mikrobiologii Instytutu Żywności i Żywienia
Kierownik Pracowni: dr n. przyr. Janusz Popowski
Summary
Mad cow disease belongs to the small group of the rare specific illnesses caused by degenerated proteins named prions. In result of their activity central nervous system turns into the spongiform structure what is followed by characteristic symptoms with fatal outcome. Creutzfeldt--Jakob disease among humans is considered to be an analogy to BSE (bovine spongiform encephalopathy). It appears in population sporadically or as hereditary disease or it may be transferred in iatrogenic way during brain surgery. Alimentary tract is also known as the gate of infection, when certain parts of infected animals are consumed. The problem of food safety is discussed and the opinion is expressed that the risk connected with beef consumption is negligible due to the interspecies barrier (between cow and man) and due to numerous counteractions undertaken by all European countries.

Wstęp
Grupa tzw. chorób prionowych obejmuje kilkanaście wyodrębnionych jednostek chorobowych. Wśród nich najbardziej znane to występująca u ludzi choroba Creutzfeldta-Jakoba (CJD) oraz atakująca zwierzęta gąbczasta encefalopatia bydła (BSE) czy scrapie u owiec. Wszystkie choroby prionowe występowały dotychczas z małą częstością i większość z nich pozostawała niezauważona przed wprowadzeniem nowoczesnych systemów monitorowania i kontroli epidemiologicznej oraz finezyjnych metod diagnostycznych. W ostatnim piętnastoleciu jedna z nich – BSE – przełamała ten schemat, powodując masowe zachorowania wśród bydła, przede wszystkim na terenie Wielkiej Brytanii, a w niewielkim stopniu także w kilku innych krajach europejskich
Może się wydawać zaskakujące, że ten medyczny, społeczny a także, dla wielu osób, ekonomiczny dramat jakim jest epidemia gąbczastej encefalopatii bydła, nazywanej potocznie chorobą szalonych krów ma także kilka pozytywnych aspektów. Za najważniejszy z nich można uznać zdecydowane zwiększenie nakładów finansowych na naukę i intensyfikację prac licznych ośrodków badawczych, których celem jest, z jednej strony poznanie tego fascynującego z biologicznego punktu widzenia zjawiska, a z drugiej strony znalezienie środków zapobiegających chorobie i jej przeniesieniu na ludzi. Innym, może mniej istotnym, lecz nie pozbawionym pozytywnych wartości aspektem, jest fakt, że ogólna wiedza społeczeństwa na temat tych, jak dotąd, rzadkich chorób, wzrosła od poziomu niemal zerowego do poziomu wiedzy, jaki ma przeciętny zjadacz chleba o wielu innych popularnych chorobach, takich jak grypa, odra czy ospa wietrzna. Prawie każdy, kto nie stroni od prasy codziennej lub mediów audiowizualnych już wie, że czynnikiem infekcyjnym w przypadku chorób prionowych nie jest bakteria ani wirus, lecz cząsteczka białka o nazwie prion. Mówiąc o czynniku infekcyjnym mamy zazwyczaj na myśli coś, co z zewnątrz wnika do naszego organizmu przez tzw. wrota zakażenia, którymi może być przewód pokarmowy, błona śluzowa układu oddechowego, czy np. miejsce uszkodzenia naskórka. W dużej mierze jest to prawda i dla prionów, lecz w wielu przypadkach zdarza się, że prion nie jest importowany do organizmu z zewnątrz, lecz powstaje samorzutnie w jego wnętrzu. Na dodatek cecha ta, przy spełnieniu pewnych warunków, bywa dziedziczna. Ta unikatowa dla chorób prionowych właściwość zostanie dokładniej wyjaśniona w dalszej części artykułu.
Mechanizm patogenezy. Skąd się biorą priony?
Ojcem nazwy „prion” jest amerykański badacz Stanley Prusiner, który za swe olbrzymie osiągnięcia w dziedzinie rozszyfrowywania natury prionu otrzymał w 1997 roku Nagrodę Nobla (1). Samą nazwę powołał do życia kilkanaście lat wcześniej, kiedy wszystko zdawało się wskazywać, że zakaźne właściwości związane są z osobliwą cząsteczką białka (ang. protein infection), a nie z chorobotwórczym drobnoustrojem. Złogi tego białka odnajdywano w mózgach zwierząt i ludzi dotkniętych gąbczastą encefalopatią. Zjawisko to było jednak nadal trudne do interpretacji, ponieważ okazało się, że nawet bardzo mała ilość tego białka pobrana od chorych zwierząt i wprowadzona do mózgu zdrowych doświadczalnych zwierząt nie tylko wywoływała u nich gąbczastą encefalopatię lecz także prowadziła do pojawienia się w mózgu złogów zakaźnego białka w ilości znacznie większej od tej, która została wprowadzona. Jak wiadomo, samo białko pozbawione materiału genetycznego nie może się rozmnażać czy powielać, powstało więc pytanie jak dochodzi do tak nieoczekiwanego efektu.
Sprawa zaczęła się powoli wyjaśniać, kiedy w genomie człowieka i wielu zwierząt znaleziono gen odpowiedzialny za produkcję białka, w zasadzie identycznego z chorobotwórczym prionem. Białko to u normalnych zdrowych osobników jest syntetyzowane w stosunkowo niewielkich ilościach i lokuje się w błonie cytoplazmatycznej komórek, przede wszystkim nerwowych, pełniąc tam jakąś bliżej nieokreśloną funkcję. W każdym razie na pewno nie jest szkodliwe. A zatem pomiędzy zdrowym fizjologicznie prawidłowym białkiem, które określono skrótem PrPC (ang. C = cellular = komórkowe) a chorobotwórczym prionem musi istnieć jakaś inna poważna różnica. Dalsze fizykochemiczne analizy doprowadziły do wniosku, że identyczność pomiędzy oboma rodzajami cząsteczek sprowadza się do tzw. budowy pierwszorzędowej czyli kolejności ułożenia podstawowych cegiełek każdego białka – aminokwasów. Tymczasem wiadomo, że zsyntetyzowany w komórce łańcuch aminokwasowy przybiera określoną strukturę drugorzędową o nazwie a-helix, przypominającą rozciągniętą nieco spiralkę grzejnika. Niekiedy jednak, zależnie od warunków i składu cząsteczki, jej drugorzędowa struktura może przybrać formę płaskiej wstążki i nazywa się wtedy sfałdowaniem typu b. Wykazano, że w zdrowym, komórkowym białku PrPC przeważa struktura typu a, natomiast w chorobotwórczych prionach dominują odcinki białka ukształtowane według typu b.
Ta niesłychanie ważna informacja stała się podstawą do stworzenia hipotezy, która mimo wielu prób jej podważenia (1, 2), w najpełniejszym jak dotąd stopniu opisuje całokształt zjawisk obserwowanych przy badaniu gąbczastych encefalopatii różnego pochodzenia. Według tej hipotezy, a właściwie już dobrze udokumentowanej doświadczalnie teorii, zakaźny prion posiada, w wyniku zmienionej konformacji, cały szereg specyficznych właściwości funkcjonalnych odróżniających go od fizjologicznego białka PrPC. Chyba najważniejsza z nich to zdolność do chwilowego łączenia się z komórkowym białkiem PrPC. Podczas tego „braterskiego uścisku” prion wymusza na białku PrPC zmianę jego struktury z formy a na formę b. W ten sposób zdrowe rodzime białko samo staje się chorobotwórczym prionem i może kontynuować dalsze akty przemiany swoich siostrzanych zdrowych cząsteczek na cząsteczki zakaźne, zapoczątkowując rodzaj biologicznej reakcji łańcuchowej. Tak więc, mimo, że komórka przez cały czas biosyntetyzuje w oparciu o swoją informację genetyczną normalne białko, to jest ono natychmiast zmieniane w priony. Na dodatek, białko to o zmienionej konformacji nie jest rozpoznawane przez systemy transportowe komórki i nie jest eksportowane do błony cytoplazmatycznej, jak to się dzieje ze zdrowym białkiem PrPC, natomiast gromadzi się wewnątrz komórki.
Dalszy scenariusz wydarzeń ciągle jeszcze stanowi przedmiot badań i dyskusji. Wydaje się, że w wyniku nagromadzenia prionów dochodzi do uaktywnienia tzw. pęcherzyków lizosomalnych wypełnionych enzymami trawiennymi. Łączą się one w duże wakuole a wreszcie pękają uwalniając enzymy, które rozpuszczają błonę komórkową i powodują wylanie zawartości komórki (3).
Inny model przewiduje włączenie kaskady reakcji prowadzących do tzw. apoptozy czyli programowanej śmierci komórki. Tak czy inaczej komórka ulega destrukcji a chorobotwórcze priony uwalniają się i zakażają inne komórki z najbliższego sąsiedztwa, co w sumie prowadzi do wytworzenia gąbczastej struktury i upośledzenia funkcji centralnego układu nerwowego (4, 5). Autorem tej teorii jest wspomniany już wyżej Stanley Prusiner z Kalifornijskiego Uniwersytetu w San Francisco.
Pozostaje otwarte pytanie jak dochodzi do pojawienia się w komórce mózgowej tego pierwszego prionu, który rozpoczyna swoją podstępną działalność. Odpowiedź może być różna w zależności od odmiany choroby, której mechanizm chcemy rozszyfrować. Tę rozmaitość rozwiązań przyjętych przez naturę spróbujemy usystematyzować w następnym rozdziale.
Ludzkie i zwierzęce choroby prionowe
Przedstawione w tabeli 1 choroby są nieźle poznane i udokumentowane. Lista ta nie wydaje się jednak zamknięta, gdyż sporadycznie pojawiają się w literaturze doniesienia o innych chorobach, których mechanizm patogenezy może opierać się na działaniu prionów. Wymagają one jednak dalszego potwierdzenia (4). Wymienione choroby, niezależnie od tego czy występują w organizmach ludzkich czy zwierzęcych posiadają szereg wspólnych cech. Wszystkie one rozwijają się w wyniku inwazji chorobotwórczych prionów, w następstwie czego dochodzi do powstania gąbczastej struktury mózgu i fizycznej oraz funkcjonalnej degeneracji centralnego układu nerwowego. Towarzyszy temu szereg objawów takich jak niezborność ruchów, drżenie, demencja, bezsenność, agresja, paraliż. We wszystkich przypadkach choroba kończy się śmiercią. Różnice między poszczególnymi chorobami polegają między innym na długości czasu inkubacji choroby, który jednak w większości przypadków trwa lata a nawet dziesiątki lat. Poza tym w zależności od choroby i rodzaju zaatakowanego organizmu obserwujemy przewagę takich czy innych wymienionych wyżej objawów lub kolejności ich wystąpienia.
Tabela 1. Gąbczaste encefalopatie mózgu u ludzi i zwierząt wywołane przez priony.39
| Nazwa choroby | Kiedy po raz pierwszy opisano | W jakim organizmie występuje |
| Choroba Creutzfeldta- Jakoba | 1920-21 | człowiek |
| Syndrom Gerstmanna- Sträusslera-Scheinkera | 1936 | człowiek |
| Kuru | 1957 | człowiek |
| Śmiertelna rodzinna bezsenność | 1992 | człowiek |
| Scrapie | 1732 | owca, koza |
| Zakaźna encefalopatia norek | 1947 | norka |
| Chroniczna wyniszczająca choroba zwierzyny płowej | 1980 | łoś, jeleń |
| Gąbczasta encefalopatia bydła - BSE | 1985 | krowa |
| Encefalopatia egzotycznych zwierząt kopytnych | 1987 | antylopy, dzikie odmiany bydła |
| Gąbczasta encefalopatia kotów | 1990 | kot domowy, kotowate z ZOO |
Spośród wymienionych czterech chorób prionowych spotykanych w populacji ludzkiej, dwie – śmiertelna rodzinna bezsenność oraz syndrom Gerstmanna-Sträusslera-Scheinkera występują niezwykle rzadko i dotychczas odnotowano w medycynie bardzo niewielką liczbę przypadków. Choroba kuru praktycznie nie istnieje z innego względu. Pojawiła się ona w latach pięćdziesiątych XX wieku w obrębie niewielkiej grupy etnicznej na Nowej Gwinei z częstością kilku procent, a jej rozprzestrzenianie wydawało się być związane z rytualnym spożywaniem mózgów zmarłych członków plemienia. Zaniechanie tego zwyczaju wymuszone odpowiednimi zarządzeniami władz spowodowało zanik choroby (2, 4).
Warto nieco dłużej zatrzymać się nad chorobą Creutzfeldta-Jakoba, rozpoznaną i scharakteryzowaną wstępnie przez dwóch niemieckich lekarzy w 1921 roku. W chwili obecnej wiadomo, że choroba ta występuje na całym świecie z częstością ok. 1 przypadku na milion mieszkańców. Tak więc, w takim kraju jak Polska powinno się odnotowywać rocznie ok. 40 przypadków CJD. W rzeczywistości, dane statystyczne dla naszego kraju podają nieco mniejsze wartości, ponieważ choroba dotyczy najczęściej osób w bardzo zaawansowanym wieku, w związku z czym możliwe są niezbyt precyzyjne oceny diagnostyczne i zaniechanie specjalistycznych badań. Przyjmuje się że ok. 80% przypadków CJD ma charakter sporadyczny. Przez to określenie rozumie się, że choroba nie jest dziedziczna, dotyczy tylko jednego członka rodziny i nie jest przekazywana na następne pokolenia. Wynika to z faktu, że w komórkach mózgowych, choć bardzo rzadko, dochodzi do spontanicznej przemiany jednej lub kilku cząsteczek białka PrPC w prion. Tak rozpoczyna się trwający wiele lat proces prowadzący w końcowym stadium do śmiertelnej choroby. Trudno powiedzieć co jest bezpośrednią przyczyną tej przemiany i czy zmiana ta zachodzi wyłącznie na poziomie białka czy jest poprzedzona jakąś mutacją w genie kodującym to białko. Hipoteza mutacji wydaje się bardzo prawdopodobna gdyż wiadomo, że pewne zmiany w budowie genu prp sprawiają, że wyprodukowane w oparciu o tę zmienioną informację genetyczną białko posiada nieco większą łatwość przyjmowania konformacji b. Należy jednak pamiętać, że jeśli wspomniana mutacja zaszła w komórce mózgowej, a więc komórce somatycznej, będzie ona dotyczyła tylko i wyłącznie osobnika, u którego wystąpiła i nie zostanie przeniesiona na następne pokolenia.
Inaczej rzecz się będzie miała wówczas, kiedy do tego typu mutacji dojdzie w komórce rozrodczej takiej jak jajo lub plemnik. Wówczas ową cechę zwiększonej podatności białka PrPC na przybieranie konformacji b otrzymają wszystkie komórki w potomstwie, a właściwość ta będzie przekazywana z pokolenia na pokolenie. Choć nie u wszystkich członków takiej rodziny choroba Creutzfeldta-Jakoba musi wystąpić, jednak prawdopodobieństwo jej będzie znacznie zwiększone. Taka odmiana choroby nosi nazwę rodzinnej CJD i stanowi ok. 15% wszystkich przypadków. Pozostałe 5% odnotowywanych w medycynie przypadków CJD stanowi jej odmiana jatrogenna, a więc powstała w trakcie niewłaściwie przeprowadzonych zabiegów medycznych wskutek bezpośredniego przeniesienia prionów z chorego na zdrowego człowieka. Takie zdarzenia miały miejsce przy przeszczepach rogówki i opony twardej, jeśli zmarły dawca zakończył życie w wyniku CJD.
Ostatnio pojawił się nowy wariant choroby Creutzfeldta-Jakoba obejmujący jak dotychczas ok. 100 osób (5, 6). Istnieją poważne podstawy by przypuszczać, że chorobotwórcze priony wtargnęły w tych przypadkach do organizmu człowieka drogą pokarmową i pochodzą z bydła zakażonego BSE.
Epidemia BSE (bovine spongiform encephalopathy) pojawiła się na Wyspach Brytyjskich w połowie lat osiemdziesiątych. O ile bezspornym wydaje się fakt, że rozprzestrzenienie choroby nastąpiło przy udziale mączki mięsno-kostnej zastosowanej jako pasza dla bydła, o tyle pochodzenie prionów w tej mączce nie jest już tak oczywiste. Do jej produkcji używa się kości i innych resztek rzeźnych owiec, bydła a także zapewne i innych zwierząt. Panuje powszechne przekonanie, że pierwotnym źródłem choroby były owce, u których od kilkuset lat znana jest choroba prionowa o nazwie scrapie. Istotnie, wykazano wprowadzając do mózgu doświadczalnych krów próbkę mózgu chorej owcy, że taka droga jest możliwa. Zakażone w ten sposób krowy chorowały na gąbczastą encefalopatię, choć przebieg choroby różnił się w szczegółach od innych przypadków BSE. Inna hipoteza mówi, że rezerwuarem choroby były same krowy, u których BSE być może występowała zawsze, lecz z tak niską częstością, że pozostawała niezauważona przez hodowców. Wystarczyło jednak, że jedna chora sztuka dostała się do łańcucha pokarmowego bydła za pośrednictwem mączki, a cykl ten mógł się powtórzyć kilkakrotnie aż choroba ujawniła się przybierając rozmiary epidemiczne. Do mniej wiarygodnych, lecz nie całkiem nieprawdopodobnych, należy pogląd iż BSE pochodzi od ludzkiej choroby Creutzfeldta-Jakoba. Pośrednim ogniwem byłyby tu krowy używane do produkcji pewnego typu przeciwciał. Były one poddawane immunizacji za pomocą ekstraktów z ludzkiej przysadki mózgowej. Dawcami gruczołów przysadkowych byli oczywiście zmarli a wśród nich mogli się trafić i tacy, których przyczyną zgonu była CJD. Bydło wykorzystane w tym procesie mogło być po skrwawieniu kierowane do dalszej utylizacji, do produkcji mączki mięsno-kostnej (7, 8).
W literaturze dotyczącej tej problematyki można znaleźć ponad dwadzieścia jeszcze innych prób wyjaśnienia pochodzenia BSE, których tu nie będziemy prezentować, ponieważ wiele z przedstawianych tam pomysłów często bliższych jest naukowej fantastyki niż nauki sensu stricto.
Wymienione w tabeli inne prionowe choroby zwierzęce nie mają charakteru epidemicznego ani nie stanowią zagrożenia dla żadnej populacji. Część z nich (np. encefalopatia kotów domowych lub zwierząt w ZOO) mogła się pojawić w wyniku zastosowania w karmie resztek rzeźnych zakażonego bydła.
Analiza ryzyka
W połowie lat osiemdziesiątych XX wieku pojawiły się w Anglii pierwsze przypadki choroby szalonych krów (BSE). W kolejnych latach liczba ich gwałtownie rosła by osiągnąć szczyt w 1992 roku. Od tego czasu liczba zachorowań wśród zwierząt spada i w tej chwili jak to pokazuje przebieg krzywej na rycinie 1, zbliża się asymptotycznie do zera. Spadek ten wynika ze zrozumienia mechanizmu przenoszenia choroby i podjęcia przez władze brytyjskie odpowiednich kroków, takich jak zakaz karmienia bydła mączką mięsno-kostną, a następnie całkowity zakaz jej produkcji, a nawet posiadania pod groźbą surowej kary. Jednocześnie doszło do eksterminacji ponad 2 milionów sztuk bydła podejrzanego o kontakt z chorymi sztukami lub karmionego mączką mięsno-kostną, nawet jeśli nie zaobserwowano u nich objawów chorobowych (8).
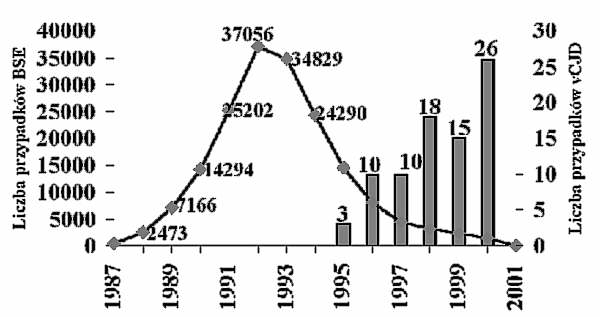
Ryc. 1. Liczba przypadków BSE i nowego wariantu choroby Creutzfeldta-Jakoba (vCJD) w Wielkiej Brytanii.41
Zanim jednak wprowadzono w życie te wszystkie restrykcyjne przepisy znaczna część zakażonego bydła znalazła się na stołach angielskich i nie tylko. Epidemiolodzy obliczają, że w samej Anglii spożyto 300 000-500 000 sztuk bydła, które mogło zawierać chorobotwórcze priony. Praktycznie więc poza ścisłymi wegetarianami nie było mieszkańca Wysp Brytyjskich, który nie skosztowałby skażonej wołowiny i to niekoniecznie tylko w postaci befsztyka lecz także w postaci pasztetów, salcesonów i wędlin, i wielu innych wyrobów, które mogły zawierać w znacznym procencie takie fragmenty zwierzęcego organizmu, które są obecnie uważane za wyjątkowo niebezpieczne (tab. 2).
Tabela 2. Kategoryzacja zakaźności narządów i tkanek zwierząt chorych na BSE wg Sterującego Komitetu Naukowego Unii Europejskiej.41
| Kategoria zakaźności | Narząd, tkanka |
| 1. Wysoka | mózgowie, oczy, rdzeń kręgowy, zwoje grzbietowe, opona twarda, przysadka, czaszka, kręgosłup, płuca |
| 2. Średniego stopnia | całe jelita, migdałki, śledziona, łożysko, macica, tkanki płodu, nadnercza, płyn mózgowo-rdzeniowy, węzły chłonne |
| 3. Niskiego stopnia | wątroba, trzustka, grasica, szpik kostny, kości, śluzówka nosa, nerwy obwodowe |
| 4. Nie wykazano | mięśnie szkieletowe, serce, nerki, siara, mleko, tkanka tłuszczowa, ślinianki, ślina, tarczyca, gruczoł mlekowy, jajniki, jądra, pęcherzyki nasienne, chrząstka, tkanka łączna, skóra, włosy, skrzep krwi, surowica, mocz, żółć, kał |
Przez dość długi okres nie istniały również żadne blokady eksportowe. Podaje się, że w latach 1985-90 wyeksportowano z Anglii ponad 50 000 sztuk bydła i trudną do określenia ilość mięsa i jego przetworów, a także tysiące ton mączki mięsno-kostnej (8). Część z tej produkcji znalazła się także w Polsce.
Dzisiaj, z perspektywy czasu, można ocenić skutki zarówno tego okresu beztroski jak i późniejszego, kiedy skojarzono już związki przyczynowo-skutkowe, dotyczące BSE i podjęto kroki zapobiegawcze.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Liberski P.: Nagroda Nobla 1997. Priony – za i przeciw kontrowersyjnej hipotezie. Post. Mikrobiol. 1999, 37, 9.– 2. Popowski J.: Priony. Post. Mikrobiol., 1993, 32, 53.– 3. Polak M.P, Żmudziński J.F.: Diagnostyka zakaźnych gąbczastych encefalopatii. Medycyna Wet. 2000, 56, 143.– 4. Deptuła W, Pawlikowska M.: Charakterystyka chorób prionowych – wybrane dane. Medycyna Wet. 2000, 56, 11.– 5. Molenda J.: Nowa odmiana choroby Creutzfeldta-Jakoba a gąbczasta encefalopatia u bydła. Medycyna Wet. 2000, 56, 355.– 6. UK Department of Health. Monthly CJD Statistical Figures – http:// www.doh.gov.uk/cjd_stat.htm.– 7. Official Mad Cow Disease Home Page – http://mad-cow.org.– 8. Żmudziński J.F, Polak M.P.: BSE – mity i fakty. Życie Wet. 2001, 76, 31.– 9. Lee H.S. et al.: Increased Susceptibility to Kuru of Carriers of the PRNP 129 Methionine/Methionine Genotype. J. Infect. Dis. 2001, 183, 192.– 10. Will R.G. et al.: Diagnosis of New Variant Creutzfeldt-Jakob disease. Ann.Neur. 2000, 47, 575.– 11. Bratosiewicz J. et al.: Molecular analysis of PRNP gene in Polish population and in Creutzfeldt-Jakob disease. Folia Nuropathol. 1999, 37, 277.– 12. National Cattlemen's Beef Association – http://www.bseinfo.org/resource/vcjd_fact.htm.– 13. Oppenheim C. et al.: MRI and the second French case of vCJD. Lancet. 2000, 356, 253.– 14. Biuletyn Informacyjny Gelatines Weishardt, Graulhet, France, 1998.– 15. Jackson G.S. et al.: Reversible conversion of monomeric human prion protein between native and fibrilogenic conformations. Science. 1999, 283, 1935.
