© Borgis - Postępy Nauk Medycznych s2/2011, s. 3-10
*Izabela Korczowska1, Paweł Hrycaj1, Jan K. Łącki2
Białka ostrej fazy we współczesnej diagnostyce medycznej**
Application of acute phase proteins in modern medical diagnostics
1Zakład Reumatologii i Immunologii Klinicznej Uniwersytetu Medycznego w Poznaniu
Kierownik Zakładu: dr hab. med. Paweł Hrycaj, prof. UM
2Katedra Zdrowia Publicznego Uniwersytetu Zielonogórskiego
Kierownik Katedry: prof. dr hab. Jan. K. Łącki
Streszczenie
Białka ostrej fazy stanowią bardzo ważne wskaźniki diagnostyczne oraz rokownicze zarówno w przebiegu zakażeń, zapalenia, chorób nowotworowych, po przeszczepach, jak i w innych stanach zaburzenia homeostazy ustroju. Priorytetowym działaniem białek ostrej fazy jest przywracanie homeostazy organizmu poprzez aktywację układu dopełniacza, reakcję nieswoistą związaną z opsonizacją i aglutynacją, ograniczaniem uszkodzeń tkanek powodowanych przez bakterie oraz enzymy lizosomalne z komórek fagocytujących oraz wzmożoną aktywność chemotaktyczną. Szczególne znaczenie wśród białek ostrej fazy zajmuje białko C-reaktywne będące najczęściej oznaczanym wskaźnikiem stanu zapalnego. Jego oznaczanie pozwala nie tylko na potwierdzenie toczącej się infekcji, ale jest również wskaźnikiem zapalenia wątroby, trzustki, chorób naczyniowych, miażdżycy i wielu innych. Oznaczanie białek fazy ostrej dzięki dużej szybkości i względnie małej cenie umożliwia szybką diagnostykę oraz ocenę stopnia zaawansowania choroby, jej rozległości i dynamiki zmian, służy również do potwierdzenia chorób o podłożu genetycznym.
Summary
Acute phase proteins are very important diagnostic and prognostic factors in infections, neoplastic diseases, after transplantation and other homeostasis disturbances. The main function of acute phase proteins is homeostatic recovery by complement activation, non-specific activation of opsonisation and agglutination, diminution of tissue damage induced by bacteria and lysosomal enzymes released from phagocytes as well as enhanced chemotaxis. A special position in acute phase proteins plays C-reactive protein, the most commonly determined inflammatory index. Determination of C-reactive protein is an index of infection, hepatitis, pancreatitis, vascular disorders, atherosclerosis and other conditions. C-reactive protein assay is a rapid and relatively non-expensive test facilitating rapid diagnosis, estimation of progress of the disease, its dynamics and involved organs as well as is applied in diagnostics of heteritary disorders.

Reakcja organizmu na pojawiający się czynnik uszkadzający struktury tkankowe lub narządowe przejawia się jako odczyn zapalny. Jest on wyrazem swoistej ukierunkowanej oraz wzmożonej odpowiedzi biochemicznej, hematologicznej i immunologicznej na poziomie lokalnym lub ogólnoustrojowym. Umiarkowany odczyn zapalny jest niezwykle korzystny dla ustroju, prowadzi bowiem do usuwania produktów martwiczych, do hamowania krwawienia powstałego podczas urazu czy też wydalania wraz z wysiękiem endo- i egzotoksyn. Pojęcie ostrej fazy wprowadzili Abernethy i Avery w 1941 r. (1) opisując właściwości osocza gorączkującego chorego, w którym stwierdzono występowanie białka C-reaktywnego. Miller i wsp. (2) w 1951 r. wykazali, że głównym miejscem syntezy białek ostrej fazy (BOF) jest wątroba. Ich synteza zachodzi jako odpowiedź na zaburzenia homeostazy wywołanej przez ostre stany zapalne, zakażenia bakteryjne, nowotwory czy też choroby autoimmunizacyjne. BOF ograniczają rozprzestrzenianie się procesu zapalnego i usuwają jego skutki. Ich priorytetowym działaniem jest przywracanie homeostazy organizmu poprzez aktywację układu dopełniacza, reakcję nieswoistą związaną z opsonizacją i aglutynacją, ograniczaniem uszkodzeń tkanek powodowanych przez bakterie oraz enzymy lizosomalne z komórek fagocytujących oraz wzmożoną aktywność chemotaktyczną (ryc. 1). Wpływają one nie tylko na działanie układu immunologicznego, ale także na wydzielanie niektórych hormonów np. glikosterydy, ACTH (3).
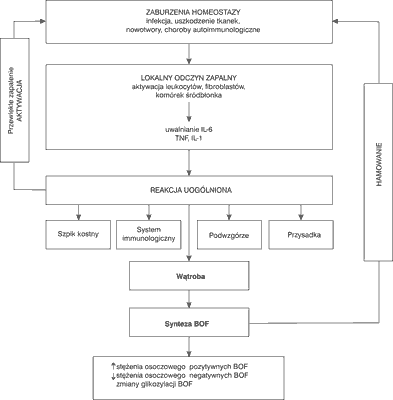
Ryc. 1. Regulacja odpowiedzi zapalnej w ustroju.
Skróty: BOF = białka ostrej fazy; IL-1 = interleukina-1; IL-6 = interleukina-6; TNF = tumor necrosis factor
Główne cytokiny odpowiedzialne za indukcję syntezy BOF to interleukina-6 (IL-6), interleukina-1 (IL-1) TNF-α i TNF-β. IL6 wpływa na fosforylację czynnika transkrypcyjnego NF-IL6, który w wyniku modyfikacji transportowany jest do jądra komórkowego, gdzie następnie aktywuje transkrypcję genów dla BOF. IL-1 oraz TNF fosforylują inhibitor czynnika transkrypcyjnego NFkB, powodując uwalnianie NFκB i aktywację genów w jądrze komorowym. Leukocyty, monocyty i makrofagi wytwarzają mediatory ostrej fazy, które stymulują hepatocyty do syntezy BOF (4). Najsilniejszym aktywatorem BOF jest IL-6, natomiast IL-1 i TNF stymulują fibroblasty, komórki endotelium, keratynocyty do syntezy IL-6 i dodatkowo wzmacniają jej biologiczną aktywność.
Podział białek ostrej fazy
Białka ostrej fazy to heterogenna grupa białek, różniąca się właściwościami fizykochemicznymi, stężeniem w osoczu, funkcjami biologicznymi i dynamiką zmiany stężeń w przebiegu zapalenia. Dlatego też spotykamy różne podziały BOF (5-8).
Jedna z klasyfikacji BOF wyróżnia:
– białka pozytywne, których stężenie wzrasta co najmniej o 25%. Do tej grupy należy większość BOF;
– białka negatywne, których stężenie maleje o około 30%. Są to m.in. transferyna, prealbumina, albumina i inter-α-antytrypsyna, α1-lipoproteina;
– białka ostrej fazy obojętne, do których zaliczana jest α2-makroglobulina, protrombina, surowiczy amyloid P. Ich stężenie zmienia się jedynie nieznacznie.
W innej klasyfikacji pod uwagę bierze się stopień wzrostu stężenia:
– grupa białek, których stężenie mo-że wzrosnąć 100-1000-krotnie to białko C-reaktywne, surowiczy składnik amyloidu A;
– białka, których stężenie wzrasta o około 50%: ceruloplazmina, składowe dopełniacza C3 i C4, α1-inhibitor proteinaz, α2-antyplazmina, C1-inaktywator;
– białka, których stężenie wzrasta 2-3 krotnie: α1-antychymotrypsyna, α1-kwaśna glikoproteina, haptoglobina, α1-antytrypsyna, ferrytyna, fibrynogen.
Ze względu na kinetykę zmian wyróżnić możemy 2 grupy:
– białka I rzutu, których stężenie wzrasta najwcześniej, po 6-8 h od momentu zadziałania bodźca inicjującego reakcję ostrej fazy. Najwyższe wartości osiąga po 24-48 h i w ciągu kilkunastu godzin ulega normalizacji. Są to białko CRP, α1-antychymotrypsyna oraz surowiczy składnik amyloidu A;
– białka II rzutu, wzrost ich stężenia następuje po 24-48 h, szczyt przypada na 72 h, eliminowane są po kilku lub kilkunastu dniach i dopiero po tym czasie ich stężenia normalizują się. Do tej grupy zalicza się pozostałe białka (nie wymienione w grupie I).
Stężenie białek ujemnych obniża się w reakcji zapalnej z powodu zwiększonego katabolizmu i mniejszej syntezy w wątrobie. W bardzo wczesnym stadium reakcji zapalnej na skutek zwiększonej przepuszczalności naczyń oraz zwiększonego katabolizmu, przejściowo dochodzi do obniżenia niektórych dodatnich BOF np. składowych dotleniacza, inhibitorów proteaz czy haptoglobiny (7, 9, 10).
Glikozylacja białek ostrej fazy
W warunkach in vivo aktywacja BOF uwarunkowana jest wzajemnymi oddziaływaniami sieci cytokin. Złożoność tej odpowiedzi warunkuje odmienne wzorce w indukcji ostrej fazy. Osobnym zagadnieniem jest glikozylacja BOF oraz zmiany jej profilu. Wiadomo, że nawet niewielka modyfikacja białka, jakim jest dołączenie łańcucha cukrowego zmienia w istotny sposób jego punkt elektryczny, rozpuszczalność, aktywność biologiczną czy też antygenowość. Większość białek ostrej fazy z wyjątkiem CRP, albuminy i surowiczego składnika amyloidu A to glikoproteiny syntetyzowane przez wątrobę. Glikozylacja BOF zachodzi w siateczce endoplazmatycznej hepatocytów. Powstają one na skutek kowalencyjnego połączenia jednej lub kilku reszt węglowodanowych, tworzących rozgałęzione łańcuchy do grupy amidowej asparaginy. Boczne łańcuchy węglowodanowe glikoprotein połączone są wiązaniem N-glikozydowym (N-acetyloglukozaminy) z asparaginą łańcucha polipeptydowego. Inne możliwości to O-glikozydowe wiązanie galaktozy z 5-hydroksylizyną lub wiązanie O-glikozydowym GlcNAc z seryną lub treoniną (11). Wszystkie złożone oligosacharydy zawierają identyczny rdzeń utworzony z dwóch reszt N-acetyloglukozoaminy i z trzech reszt mannozy oraz dwa, trzy lub cztery łańcuchy boczne, nazwane „antenami”. Anteny te zbudowane są głównie z N-acetyloglukozoaminy, galaktozy i galaktozoaminy. Różnica w strukturze łańcuchów cukrowych określana jest pojęciem mikroheterogenności, która obejmuje formę główną i formę poboczną. Mikroheterogenność główna dotyczy różnorodności struktur antenarnych, poboczna natomiast różnic ilościowych kwasu sjalowego i fukozy. W przebiegu stanów zapalnych obserwuje się zaburzenie proporcji występujących fizjologicznie wariantów, często pojawiają się inne o zupełnie zmienionej reakcji z daną lektyną.
α1-kwaśna glikoproteina (AGP) jest najbardziej znanym przedstawicielem tej grupy. Badania Bierhuizena i wsp. (12) przeprowadzone z zastosowaniem chromatografii powinowactwa z lektyną – konkanawaliną A (ConA) wykazały obecność trzech frakcji AGP w surowicy. Glikoproteiny AGP-A nie wiązały się do ConA i zawierały mniej więcej równe ilości troj- i czteroantenarnych łańcuchów węglowodanowych. Natomiast frakcje AGP-B i AGP-C zawierały odpowiednio jeden lub dwa łańcuchy dwuantenarne i zbliżone ilości łańcuchów trój- i czteroantenarnych, dołączonych do pozostałych cząsteczek asparaginy łańcucha polipeptydowego. Całkowita masa cząsteczkowa AGP wynosi około 41 000, w tym 45% stanowią węglowodanowe łańcuchy boczne. Przy zastosowaniu immunoelekroforezy powinowactwa z ConA wykrywa się cztery warianty glikozylacji AGP:
1. Wariant 0 – glikoproteiny nie reagujące z ConA, posiadające wyłącznie trój- lub czteroantenarne łańcuchy węglowodanowe, dołączone do łańcucha polipeptydowego.
2. Wariant 1 – to glikoproteiny słabo reagujące z ConA, posiadające 1 łańcuch dwuantenarny.
3. Wariant 2, glikoproteiny reagujące z ConA, posiadające 2 dwuantenarne łańcuchy.
4. Dodatkowo wariant 3 – AGP, który silnie reaguje z ConA, pojawiający się w przebiegu zapaleń ostrych (wykrywany tylko przy zastosowaniu immunoelekroforezy powinowactwa z ConA).
Proporcje składowych mające różne struktury antenowe w stanach patologicznych ulegają charakterystycznym zmianom (13). Przewlekłe stany zapalne (zakażenia bakteryjne, zapalenie wątroby, reumatoidalne zapalenie stawów, zesztywniające zapalenie stawów i inne) prowadzą do zwiększonej syntezy wariantu 1. Ostre zapalenia np. zapalenie trzustki, ostre infekcje bakteryjne, zawał serca czy też uraz prowadzą do zwiększonej syntezy ConA reaktywnych form AGP. W przebiegu tocznia rumieniowatego układowego oznaczanie profilu glikozylacji BOF pomaga różnicować zaostrzenie choroby (wskaźnik AGP-RC jest prawidłowy i wynosi 1,28 ± 0,25) od wystąpienia wtórnych zakażeń bakteryjnych (wskaźnik jest znacznie podwyższony RC < 1,53) (ryc. 2). Profil glikozylacji BOF może być również wykorzystywany do diagnostyki różnicowej chorób reumatycznych; zapalenie wielomięśniowe a polimialgia reumatyczna (PM): obniżone wartości AGP-RC i ACT-RC w PM oraz podwyższone AGP-RC i ACT-RC w przebiegu zapalenia wielomięśniowego oraz do oceny aktywności choroby w sarkoidozie, w aktywnym zapaleniu zauważono wyraźne obniżenie współczynników AGP-RC i ACT-RC przy prawidłowym CRP (14, 15).
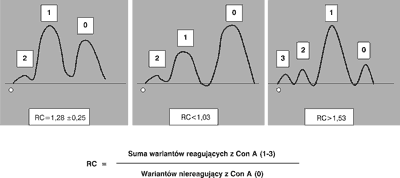
Ryc. 2. Zmiany glikozylacji AGP. Wariant 0: nie reagujące z konkanawaliną A, wariant 1: słabo reagujący z konkanawaliną A, wariant 2: silnie reagujący z konkanawaliną A, wariant 3: bardzo silnie reagujący z konkanawaliną A
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Abernethy TJ, Avery OT: The occurrence during acute infections of a protein not normally present in the blood: Distribution of the reactive protein in patients’ sera and the effect of calcium on the flocculation reaction with c polysaccharide of pneumococcus. J Exp Med 1941; 31: 173-182.
2. Miller Ll, Bly Cg, Watson Ml et al.: The dominant role of the liver in plasma protein synthesis: a direct study of the isolated perfused rat liver with the aid of lysine-epsilon-C14. J Exp Med 1951; 94: 431-453.
3. Heinrich PC, Castell JV, Andus T: Interleukin-6 and the acute phase response. Biochem J 1990; 265: 621-636.
4. Govinden R, Bhoola KD: Genealogy, expression, and cellular function of transforming growth factor-ß. Pharmacol Ther 2003; 98: 257-265.
5. Morley JJ, Kusher I: Serum C-reactive protein levels in disease. Ann NY Acad Sci 1982; 389: 406-418.
6. Gabay C, Kusher I: Acute phase protein and other systemic responses to inflammation. N Eng J Med 1999; 340: 448-454.
7. Koj A: Biological functions of acute phase proteins. [In:] The acute phase response to injury and infection. Gordon AH, Koj A, [eds.], Elsevier, Amsterdam, New York, Oxford 1985; 145-160.
8. Koj A, Kurdowska A, Magielska-Zero D et al.: Limited effects of recombinant human and murine interleukin 1 and tumour necrosis factor on production of acute phase proteins by cultured rat hepatocytes. Biochem Int 1987; 14: 553-556.
9. Esmon CT: The impact of the inflammatory response on coagulation. Thromb Res 2004; 114: 321-327.
10. Beaudeux JL, Giral P, Bruckert E et al.: Matrix metalloproteinases, inflammation and atherosclerosis: therapeutic perspectives. Clin Chem Lab Med 2004; 42: 121-131.
11. Mackiewicz A, Kushne I: Affinity electrophoresis for studies of mechanisms regulating glycosylation of plasma proteins. Electrophoresis 1989; 10: 830-835.
12 Bierhuizen MF, De Wit M, Govers CA et al.: Glycosylation of three molecular forms of human alpha 1-acid glycoprotein having different interactions with concanavalin A. Variations in the occurrence of di-, tri-, and tetraantennary glycans and the degree of sialylation. Eur J Biochem 1988; 175: 387-394.
13. Lacki JK, Sobieska M, Leszczynski P et al.: Microheterogeneity of alpha 1-acid glycoprotein in patients with rheumatoid arthritis. Ann Rheum Dis 1994; 53: 480.
14. Lacki JK, Klama K, Samborski W et al.: Comparison of microheterogeneity of alpha-1-acid-glycoprotein in serum and synovial fluid from rheumatoid arthritis patients. Clin Rheumatol 1994; 4: 598-604.
15. Łącki JK, Korczowska I: Immunopatologia chorób tkanki łącznej. [W:] Objawy reumatyczne w przebiegu chorób narządów wewnętrznych: Pazdur J. [red.], Termedia Poznań 2010; 19-33.
16. Hrycaj P: Microheterogeneity of alpha 1-antitrypsin in relation to the concentration of its complex with immunoglobulin A in the sera of patients with rheumatoid arthritis. Clin Exper Rheumatol 1996; 14: 119-123.
17. Tillett WS, Francis Tjr: Serological reactions in pneumonia with a nonprotein somatic fraction of pneumococcus. J Exp Med 1930; 52: 561-585.
18. Blake GJ, Ridker PM: Inflammatory bio-markers and cardiovascular risk prediction. J Intern Med 2002; 252: 283-294.
19. Morkowski J, Wiktorowicz K, Mackiewicz S: Pentaksyny – struktura i funkcja. Post Hig Med Dośw 1987; 41: 433-452.
20. Osmand AP, Friedenson B, Gewurz H et al.: Characterization of C-reactive protein and the complement subcomponent C1t as homologous proteins displaying cyclic pentameric symmetry (pentraxins). Proc Natl Acad Sci USA 1977; 74: 739-743.
21. Baumann H, Gauldie J: The acute phase response. Immunol Today 1994; 15, 74-80.
22. Heegaard NHH, Robe FA: A capillary electrophoresis-based assay for the binding of Ca2+ and phosphorylcholine to human C-reactive protein. J Immunol Methods 1993; 5: 103-110.
23. Munoz LE, Gaipl US, Franz S et al.: Systemic lupus erythematosus – a disease of clearance deficiency. Rheumatology (Oxford) 2005; 44: 1101-1107.
24. Volanakis JE: Human C-reactive protein: expression, structure, and function. Mol Immunol 2001; 38: 189-197.
25. Bell SA, Du Clos TW, Khursigara G et al.: Autoantibodies to cryptic epitopes of C-reactive protein and other acute phase proteins in the toxic oil syndrome. J Autoimmun 1995; 8: 293-303.
26. Rosenau BJ, Schur PH: Antibodies to C-reactive protein. Ann Rheum Dis 2006; 65: 674-676.
27. Robey FA, Jones KD, Steinberg AD: C-reactive protein mediates the solubilization of nuclear DNA by complement in vitro. J Exp Med 1985; 161: 1344-1356.
28. Bell SA, Faust H, Schmid A et al.: Autoantibodies to C-reactive protein and other acute-phase proteins in systemic autoimmune diseases. Clin Exp Immunol 1998; 113: 327-332.
29. Sjöwall C, Eriksson P, Almer S et al.: Autoantibodies to C-reactive protein is a common finding in SLE, but not in primary Sjögren’s syndrome, rheumatoid arthritis or inflammatory bowel disease. J Autoimmun 2002; 19: 155-160.
30. Sjöwall C, Bengtsson A, Sturfelt G et al.: Serum levels of autoantibodies against monomeric C-reactive protein are correlated with disease activity in systemic lupus erythematosus. Arthritis Res Ther 2004; 6: 87-94.
31. Korczowska-Łącka I, Cieślak D, Retman M et al.: Przeciwciała przeciwko białku C-reaktywnemu u chorych na toczeń rumieniowaty układowy. Reumatologia 2009; 47: 136-142.
32. Kovacs A, Green F, Hansson LO et al.: A novel common single nucleotide polymorphism in the promoter region of the C-reactive protein gene associated with the plasma concentration of C-reactive protein. Atherosclerosis 2005; 178: 193-198.
33. Szalai AJ, Wu J, Lange EM et al.: Single-nucleotide polymorphisms in the C-reactive protein (CRP) gene promoter that affect transcription factor binding, alter transcriptional activity, and associate with differences in baseline serum CRP level. J Mol Med 2005; 83: 440-447.
34. Lankisch PG: The problem of diagnosing chronic pancreatitis. Dig Liver Dis 2003; 35: 131-134.
35. Soetiono S, Sunartini, Machfudz S et al.: Cerebrospinal fluid C-reactive protein in the diagnosis of meningitis in children. Paediatr Indones 1989; 29: 20-27.
36. Szymczak E, Laskowska-Klita T, Helwich E et al.: Ocena przydatności oznaczania wybranych białek ostrej fazy we wczesnym rozpoznaniu zakażeń bakteryjnych u noworodków urodzonych przedwcześnie. Ped Pol 2000;75: 63-69.
37. Heeschen C, Hamm CW, Bruemmer J et al.: Predictive value of C-reactive protein and troponin T in patients with unstable angina: a comparative analysis. J Am Coll Cardiol 2000; 35: 1535-1542.
38. Ridker PM, Rifai N, Stampfer MJ et al.: Plasma concentration of interleukin-6 and the risk of future myocardial infarction among apparently healthy men. Circulation 2000; 101: 1767-1772.
39. Boncler M, Luzak B, Watała C: Znaczenie białka C-reaktywnego w patofizjologii miażdżycy. Post Hig Med Dośw 2006; 60: 538-546.
40. Torzewski J, Torzewski M, Bowyer DE et al.: C-reactive protein frequently colocalizes with the terminal complement complex in the intima of early atherosclerotic lesions of human coronary arteries. Arterioscler Thromb Vasc Biol 1998; 18: 1386-1392.
41. Calabro P, Willerson JT, Yeh ET: Inflammatory cytokines stimulated C-reactive protein production by human coronary artery smooth muscle cells. Circulation 2003; 108: 1930-1932.
42. Ciubotaru I, Potempa LA, Wander RC: Production of modified C-reactive protein in U937-derived macrophages. Exp Biol Med (Maywood) 2005; 230: 762-770.
43. Emerging Risk Factors Collaboration, Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J: C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet 2010; 375:132-140.
44. Gartner JC, Zitelli BJ, Malatack JJ et al.: Orthotopic liver transplantation in children: two year experience with 47 patients. Pediatrics 1984; 74: 140.
45. Migliazza L, Lopez Santamaria M, Murzia J et al.: Long-term survival expectancy after liver transplantation in children. J Pediatr Surg 2000; 35: 5-8.
46. Bakuła A, Socha P: Postać wątrobowa niedoboru alfa-1-antytrypsyny. Post Nauk Med 2010; 21: 51-57
47. Thomas GR, Forbes JR, Roberts EA et al.: The Wilson disease gene: spectrum of mutations and their consequences. Nature Genet 1995; 9:210-217.
48. Gollan JL, Gollan TJ: Wilson disease in 1998: genetic, diagnostic and therapeutic aspects. J Hepatol 1998; 28: 28-36.
49. Ferenci P, Caca K, Loudianos G et al.: Diagnosis and phenotypic classification of Wilson disease. Liver Int 2003; 23: 139-142
