© Borgis - Nowa Pediatria 4/2013, s. 155-158
*Zbigniew Krenc1, 2, Sylwia Kowalczyk-Jurgiel3
Zespół wydłużonego odstępu QT u 13-letniej dziewczynki z nawracającymi zasłabnięciami o podłożu ortostatycznym – opis przypadku
Long QT syndrome in 13-year-old girl with recurrent orthostatic presyncope – case report
1Pracownia Kardiologii Sportowej, Klinika Pediatrii, Kardiologii Prewencyjnej i Immunologii Wieku Rozwojowego, Uniwersytet Medyczny, Łódź
Kierownik Pracowni: dr n. med. Zbigniew Krenc
Kierownik Kliniki: prof. dr hab. n. med. Krzysztof Zeman
2Klinika Pediatrii i Immunologii z Pododdziałem Nefrologii, Instytut Centrum Zdrowia Matki Polki, Łódź
Kierownik Kliniki: prof. dr hab. n. med. Krzysztof Zeman
3NZOZ – Medycyna Sportowa „Twoje Zdrowie”, Łódź
Dyrektor NZOZ: dr Krzysztof Jurgiel
Summary
Syncope occurs commonly in children and adolescents. Such an event may result from a wide variety of causes, ranging from benign conditions to life-threatening diseases.
Cardiogenic syncope are rare causes of loss of consciousness and indicate on serious pathology in the cardiovascular system, such as cardiomyopathies, e.g. hypertrophic cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy, and ion channelopathies, e.g. Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia or long QT syndrome.
Congenital long QT syndrome is a genetic condition characterized by a prolonged QT interval on the surface ECGand is associated with a high risk of sudden cardiac death due to ventricular tachyarrhythmias. Mutations within 13 identified genes cause a variety of channelopathies affecting myocardial repolarization, resulting in QT prolongation.
Physical activity is not per se the cause of the enhanced mortality in long QT syndrome but it rather acts as a trigger of cardiac arrest. So, pre-participation cardiovascular evaluation, including a resting 12-lead ECG, allow to detect still asymptomatic athletes with life-threatening heart diseases to protect them from sudden cardiac death.
Our paper presents a case of 13-year-old female athlete with clinically asymptomatic long QT syndrome and recurrent orthostatic presyncope. Long QT syndrome was diagnosed in patient’s father too.

Wstęp
Zasłabnięcia i omdlenia są często spotykanym objawem w populacji dzieci i młodzieży, a także równie częstym powodem zgłaszania się do oddziałów pomocy doraźnej oraz przyczyną hospitalizacji (1).
Zazwyczaj mają podłoże czynnościowe wynikające z zaburzonych mechanizmów autoregulacji układu krążenia w zakresie utrzymania właściwego oporu oraz pojemności łożyska naczyniowego, a tym samym zabezpieczenia prawidłowej perfuzji mózgu.
Objawy te zwykle towarzyszą długotrwałej pionizacji (czasem też długotrwałej pozycji siedzącej), szybkim zmianom pozycji ciała, nierzadko występują także bezpośrednio po zakończonym wysiłku fizyczny. Dotyczą najczęściej dzieci powyżej 12 roku życia. Częściej występują u płci żeńskiej (2).
W diagnostyce różnicowej stanów przebiegających z nagłą utratą przytomności należy uwzględnić także przyczyny kardiologiczne, nierzadko stanowiące potencjalne zagrożenie nagłą śmiercią sercową. Należą do nich organiczne choroby serca, jak np. kardiomiopatia przerostowa czy arytmogenna kardiomiopatia prawej komory, ale także genetycznie uwarunkowane choroby kanałów jonowych, jak zespoły wydłużonego i skróconego odstępu QT, zespół Brugadów czy polimorficzny katechoalminergiczny częstoskurcz komorowy (3).
Niniejsza praca prezentuje przypadek 13-letniej dziewczynki z nawracającymi zasłabnięciami o ortostatycznym podłożu oraz rozpoznanym w trakcie prowadzonej diagnostyki kardiologicznej, klinicznie bezobjawowym, zespołem wydłużonego odstępu QT (LQTS).
Opis przypadku
Dziewczynka 13-letnia, uprawiająca od 6 miesięcy siatkówkę, skierowana została przez lekarza medycyny sportowej do Kliniki Pediatrii z powodu nawracających zasłabnięć (uczucie nagłego osłabienia, „mroczki przed oczami”) tuż po zakończonym wysiłku fizycznym. Z wywiadu również wiadomo, że pacjentka miewała często objawy przedomdleniowe związane ze zmianą pozycji ciała, zwłaszcza z szybką pionizacją. Nigdy jednak nie doszło do pełnej utraty przytomności.
W wykonanych badaniach laboratoryjnych, w tym w zakresie jonogramu, hormonów tarczycy, nie stwierdzono odchyleń od normy.
Próba ortostatyczna czynna (10 minut pionizacji poprzedzone 10 minutami leżenia) wykazała zaburzoną reakcję sercowo-naczyniową w odpowiedzi na pionizację, przebiegającą ze spadkiem ciśnienia (Δmax. -33 mmHg) i rozwijającą się tachykardią ortostatyczną (Δmax.+74/min).
Od 8 minuty pionizacji pacjentka zgłaszała miernie nasilone objawy przedomdleniowe.
Spoczynkowe badanie EKG wykazało cechy niemiarowości oddechowej z czynnością serca 55-64/min oraz wydłużony odstęp QT o długości 500 ms (po korekcji wg wzoru Bazetta – 490 ms). Nie stwierdzono zmian w zakresie morfologii zespołu ST-T (ryc. 1).
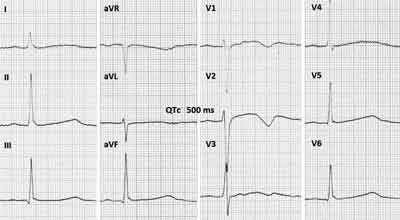
Ryc. 1. Spoczynkowe EKG u dziewczynki z zespołem wydłużonego odstępu QT.
Obserwowane zmiany w elektrokardiogramie spoczynkowym stanowiły wskazanie do poszerzenia diagnostyki kardiologicznej o całodobowe badanie EKG metodą Holtera oraz elektrokardiograficzną próbę wysiłkową.
W całodobowej rejestracji EKG metodą Holtera obserwowano rytm prowadzący zatokowy o średniej częstości 80/min z zachowaną niemiarowością oddechową, z minimalną częstością rytmu serca 46/min o godz. 4:33 i maksymalną – 149/min o godz. 16:50. Zarejestrowano 19 pojedynczych pobudzeń przedwczesnych nadkomorowych. Czas trwania niemal 94% z wszystkich odstępów QTc przekraczał wartość 470 ms, w tym aż 71% było powyżej 510 ms (manualnie obliczony odstęp QTc mieścił się w przedziale od 484 do 640 ms) (ryc. 2 i 3).
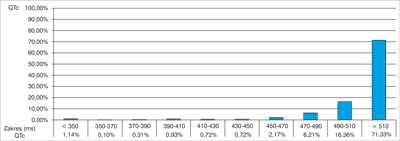
Ryc. 2. Rozkład czasów trwania odstępów QTc z pomiarów automatycznych w czasie całodobowej rejestracji EKG.

Ryc. 3. Manualne pomiary odstępów QTc w zapisie holterowskim w różnych okresach rejestracji.
Elektrokardiograficzna próba wysiłkowa, przeprowadzona na cykloergometrze rowerowym, ze stopniowo zwiększanym obciążeniem (od 25 do 75 W) wykazała prawidłową tolerancję wysiłku, nieco poniżej przeciętnej (mierzoną parametrem PWC170) wydolność fizyczną oraz prawidłową reakcję tensyjną i chronotropową na zastosowane obciążenia.
Osiągnięto maksymalną czynność serca 178/min (86% HRmax, gdzie HRmax = 220 - wiek) przy obciążeniu 75W.
Zaburzeń rytmu serca i zaburzeń przewodzenia w czasie próby nie obserwowano, a odstęp QT ulegał skróceniu wraz ze wzrostem czynności serca.
Badanie echokardiograficzne, poza łagodnymi niedomykalnościami zastawek pnia płucnego i zastawki trójdzielnej nie ujawniło istotnych zmian w zakresie budowy i czynności serca (AoD 2,3 cm; LAD 2,9 cm; RVEDD 2,18 cm; IVSD 0,79 cm; LVEDD/LVESD 4,6/3,09 cm; LVPW 0,71 cm; EF 69,7% SF% 32,8; przepływy: zastawka pnia płucnego 0,85 m/s + ślad IP; zastawka trójdzielna 0,65 m/s + ślad IT; zastawka mitralna 0,95 m/s; zastawka aortalna 1,2 m/s; aorta zstępująca 1,3 m/s).
Także konsultacje neurologiczna i okulistyczna nie ujawniły u pacjentki zmian chorobowych w zakresie OUN i narządu wzroku.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Driscoll DJ, Jacobson SJ, Porter CJ, Wollan PC: Syncope in children and adolescents. J Am Coll Cardiol 1997; 29: 1039-1045. 2. Kang MH, Xu Y, Wang C et al.: Causes of unexplained syncope in children. Zhongguo Dang Dai Er Ke Za Zhi 2012; 14(10): 771-774. 3. Farwell D, Gollob MH: Electrical heart disease: Genetic and molecular basis of cardiac arrhythmias in normal structural hearts. Can J Cardiol 2007; 23 (suppl. A): 16A-22A. 4. Meissner FL: Taubstummheit und Taubstummenbildung: Beobachtungen und Erfahrungen. Leipzig und Heidelberg: C.F. Winter’sche Verslagehandlung 1856: 119-120. 5. Jervell A, Lange-Nielsen F: Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am Heart J 1957; 54: 59-68. 6. Romano C, Gemme G, Pongiglione R: Aritmie cardiache rare dell’ eta pediatrica. Clin Pediat (Bologna) 1963; 45: 656-683. 7. Ward DC: A new familial cardiac syndrome in children. J Irish Med Assoc 1964; 54: 103-106. 8. Goldenberg I, Zareba W, Moss AJ: Long QT syndrome. Curr Probl Cardiol 2008; 33: 629-694. 9. Spazzolini C, Mullally J, Moss AJ et al.: Clinical Implications for Patients with Long QT Syndrome Who Experience a Cardiac Event During Infancy. J Am Coll Cardiol 2009; 54(9): 832-837. 10. Schwartz PJ, Moss AJ, Vincent GM, Crampton RS: Diagnostic criteria for the long QT syndrome. An update. Circulation 1993; 88: 782-784. 11. Priori SG, Schwartz PJ, Napolitano C et al.: Risk Stratification in the Long-QT Syndrome. N Engl J Med 2003; 348: 1866-1874. 12. Moss AJ, Zareba W, Schwarz KQ et al.: Ranolazine shortens repolarization in patients with sustained inward sodium current due to type-3 long-QT syndrome. J Cardiovasc Electrophysiol 2008; 19(12): 1289-1293. 13. Crotti L, Celano G, Dagradi F, Schwartz PJ: Congenital long QT syndrome. Orphanet J Rare Dis 2008; 7(3): 18.
