Dominika Drąg, Urszula Szuba, *Agnieszka Oronowicz-Jaśkowiak, Magdalena Chojnowska, Wojciech Feleszko
Zmiany w układzie oddechowym występujące u pacjentów z pierwotnymi niedoborami odporności o typie humoralnym
Changes in respiratory system in patients with primary antibody deficiencies
Klinika Pneumonologii i Alergologii Wieku Dziecięcego, Warszawski Uniwersytet Medyczny
Kierownik Kliniki: prof. dr hab. n. med. Marek Kulus
Summary
Primary antibody deficiencies are the most common primary immunodeficiencies. The diseases affect primarily pediatric patients, and their consequences extend to the whole lifespan of the individual. This group of diseases includes, among others, common variable immunodeficiency, X-linked agammaglobulinemia, selective IgA deficiency, IgM deficiency, and IgG subclass deficiency. Due to the relative rarity of these disorders and symptoms that are mostly non-specific, the diagnosis is often delayed, even by a few years. The pulmonary and respiratory changes are relatively common and include not only infectious diseases, with pneumonia being one of the most frequent complication, but also bronchiectasis, chronic lung disease, bronchopulmonary dysplasia, granulomatous-lymphocytic interstitial lung disease, and various lung tumors. The aim of this review was to present the respiratory signs and symptoms in patients with primary antibody deficiencies and discuss the therapeutic options. It must be underlined that the presence of diagnosis delay can frequently worsen the outcomes of the treatment, therefore these patients should be diagnosed as soon as it is possible.

Wstęp
Humoralne niedobory odporności, nazywane też niedoborami odporności z przewagą niedoboru przeciwciał, spowodowane nieprawidłowościami limfocytów B są najczęstszymi wśród pierwotnych niedoborów odporności i stanowią ok. 70-80% wszystkich niedoborów (1). Należą do nich m.in.: pospolity zmienny niedobór odporności (CVID), agammaglobulinemia sprzężona z chromosomem X (XLA), izolowany niedobór IgA, niedobór IgM, niedobory podklas IgG. Ze względu na rzadkie występowanie tych chorób oraz mało charakterystyczne objawy, ich rozpoznanie bywa opóźnione nawet o kilkanaście lat (2). W pracy omówiono symptomatologię powyższych niedoborów, skupiając się na manifestacjach płucnych. W zależności od różnych źródeł obejmują one od 42% (1) do 58% (3) pacjentów, a wśród nich najważniejszą grupę stanowią nawracające zakażenia górnych i dolnych dróg oddechowych wywołane bakteriami otoczkowymi (4).
Agammaglobulinemia sprzężona z chromosomem X (XLA) (agammaglobulinemia Brutona)
Ta pierwotna agammaglobulinemia spowodowana jest mutacją genu kodującego kinazę tyrozynową BTK, co skutkuje całkowitym brakiem immunoglobulin wszystkich podklas oraz śladową ilością limfocytów B (5).
U większości z pacjentów rozpoznanie zostaje postawione już w pierwszych latach życia (z reguły w 2.-3. r.ż.) (5, 6). Początkowym objawem prowadzącym do pogłębienia diagnostyki immunologicznej jest zwiększona podatność na infekcje, w szczególności: zapalenia ucha środkowego, zapalenia płuc, zapalenia zatok przynosowych, rzadziej ostre biegunki, zapalenia spojówek i tkanki łącznej (6). Najczęstszymi czynnikami etiologicznymi zapaleń płuc są bakterie otoczkowe Streptococcus pneumoniae, Haemophilus influenzae, Pseudomonas aeruginosa oraz Staphylococcus aureus (6). Pacjenci z XLA są również szczególnie podatni na enterowirusowe zakażenia ośrodkowego układu nerwowego (7).
Najczęstszym powikłaniem płucnym pacjentów z XLA jest rozstrzenie oskrzeli, zwłaszcza płatów dolnych i płata środkowego (8, 9).
Leczenie
Dożylna suplementacja immunoglobulin (IVIG) skutkuje niższą częstością infekcji i hospitalizacji (10), natomiast nie zabezpiecza przed rozwinięciem przewlekłej choroby płuc (5).
Pospolity zmienny niedobór odporności (CVID)
Pospolity zmienny niedobór odporności (CVID) to pierwotna hipogammaglobulinemia charakteryzująca się nieprawidłowym formowaniem przeciwciał z towarzyszącym obniżonym stężeniem IgG i IgA oraz, w połowie przypadków, IgM (11). Występuje z częstością 1:10 000-1:50 000 urodzeń (5, 11).
Objawy pojawiają się najczęściej przed 10. rokiem życia (2). Zwraca uwagę długi czas od wystąpienia pierwszych manifestacji klinicznych do ustalenia rozpoznania, który wynosić może nawet 10 lat (2).
Nawracające infekcje górnych i dolnych dróg oddechowych są najbardziej charakterystycznym objawem w momencie diagnozy (2). U chorych z CVID obserwuje się częstsze występowanie nowotworów układu pokarmowego i chłoniaków – ryzyko rozwoju chłoniaka u pacjentów z CVID wzrasta ponad 300-krotnie (8), a także chorób autoimmunizacyjnych, takich jak pierwotna małopłytkowość immunologiczna i niedokrwistość autoimmunohemolityczna (12).
Nawracające zakażenia układu oddechowego są dominującą symptomatologią – około połowa pacjentów przebyła zapalenie płuc co najmniej raz w okresie przed postawieniem diagnozy (2). Dodatkowo są to zapalenia zatok obocznych nosa oraz ostre zapalenie ucha środkowego wywoływane przez różne bakterie, m.in. Haemophilus influenzae (8, 13), choć obserwuje się też zwiększoną częstość zakażeń wirusowych dróg oddechowych oraz ich cięższy przebieg (8).
Astma często współistnieje z CVID, ale nadal nie ma zgody co do tego, czy jest ona skutkiem choroby, czy też schorzeniem współwystępującym (14). Istnieje hipoteza, że u części pacjentów z astmą istnieje nierozpoznany niedobór odporności, który skutkuje zwiększoną podatnością na choroby infekcyjne (15).
Rozstrzenie oskrzeli jest jednym z markerów klinicznych prowadzących do rozpoznania CVID (ryc. 1) (16). Jego obecność jest zjawiskiem odosobnionym i nie wykazuje korelacji z innymi komplikacjami płucnymi w CVID (5). Rozstrzenie oskrzeli może też być wynikiem bardzo zaawansowanych postaci śródmiąższowych chorób płuc (ang. interstitial lung disease – ILD), w trakcie których dochodzi do pociągania bliznowaciejącej i włókniejącej tkanki płucnej (17). Rozstrzenie oskrzeli z okołooskrzelowym pogrubieniem ścian najczęściej lokalizuje się w oskrzelach do dolnych segmentów płuca prawego i lewego (18). Prawdopodobnie pierwotny defekt odporności dodatkowo moduluje postęp choroby, gdyż niższa liczba limfocytów CD4+ sprzyja powstawaniu rozstrzeni (17). Mimo że rozstrzenie oskrzeli rozwija około 50% chorych, to rzadko kiedy ulegają one progresji w kierunku choroby śródmiąższowej (ILD) (19).
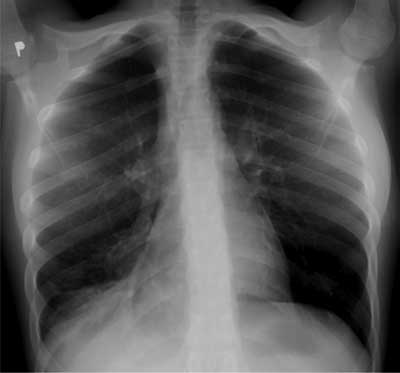
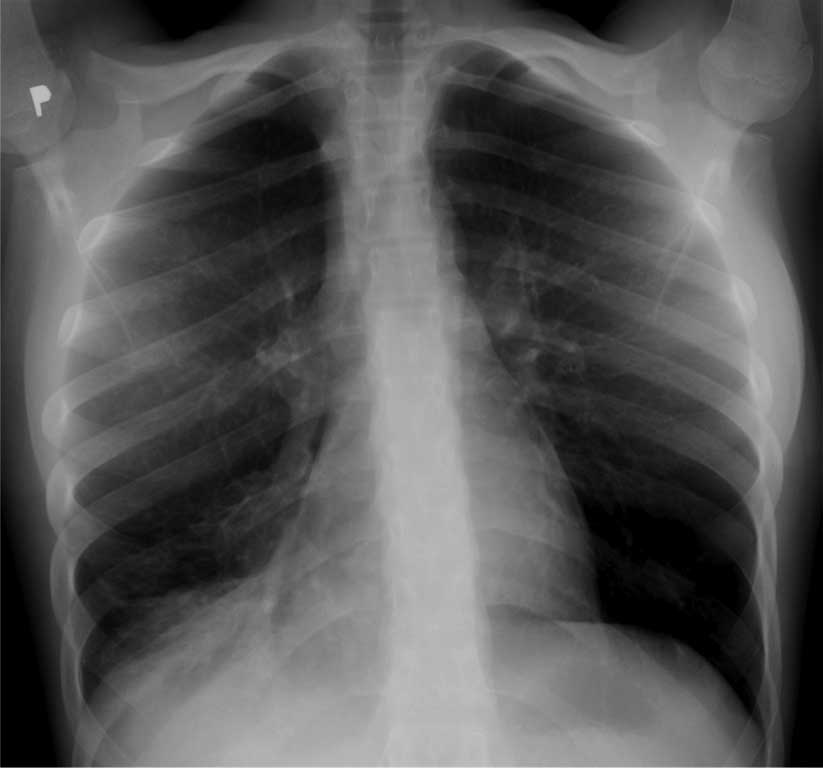
Ryc. 1. Zapalenie płuc z niedodmą płatową u 13-letniego dziecka z przewlekłymi zmianami osłuchowymi, leczonymi jako astma. U dziecka rozpoznano rozstrzenie oskrzeli jako manifestację CVID
Śródmiąższowa choroba płuc (ILD) występuje u znaczącego odsetka pacjentów z CVID (29-58%) (17). Najczęściej manifestuje się ona pod postacią: limfoidalnej hiperplazji płucnej (ang. pulmonary lymphoid hyperplasia – PLH), z grudkowym zapaleniem oskrzelików, limfocytarnym zapaleniem płuc (ang. lymphocytic interstitial pneumonitis – LIP) oraz guzkową hiperplazją limfoidalną, której manifestacją są guzki płucne (19). Patogeneza zmian płucnych w przebiegu CVID nie jest do końca wyjaśniona i nie można powiedzieć, iż zmiany są skutkiem powikłań infekcyjnych, natomiast występowanie ILD koreluje z obniżonym stosunkiem limfocytów T CD4:CD8 w popłuczynach BAL (19). Typowe objawy w tomografii komputerowej obejmują guzki, pogrubienie przegród, obraz typu mlecznej szyby oraz siateczki (20).
Przewlekła śródmiąższowa choroba płuc pozostaje wyzwaniem terapeutycznym u pacjentów z CVID, ponieważ objawy tej choroby nie ulegają wycofaniu pod wpływem substytucji IVIG (21), mimo że terapia może wiązać się z poprawą parametrów spirometrycznych (22).
Niekiedy pierwszą manifestacją CVID może być odmiana ILD, zwana ziarniniakowato-limfocytarną śródmiąższową chorobą płuc (ang. granulomatous-lymphocytic interstitial lung disease – GLILD), gdzie dominują zmiany o charakterze ziarniniaków z grudkowym zapaleniem oskrzelików, guzkowej hiperplazji limfoidalnej oraz limfocytarnego śródmiąższowego zapalenia płuc (LIP) z towarzyszącymi ziarniniakami w innych tkankach (8, 23). GLILD występuje u około 15% pacjentów z CVID (13). Rozpoznanie kliniczne stawia się na podstawie obrazu płuc w tomografii komputerowej (ryc. 2) i dowodów na obecność ziarniniaków (24). Klinicznie GLILD wiąże się z obecnością u chorych duszności, splenomegalii oraz restrykcyjnych zaburzeń oddychania z niskimi wartościami DLCO (25).
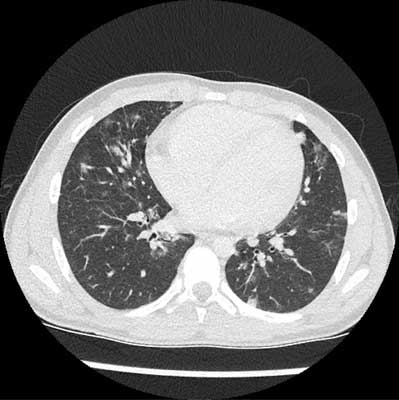
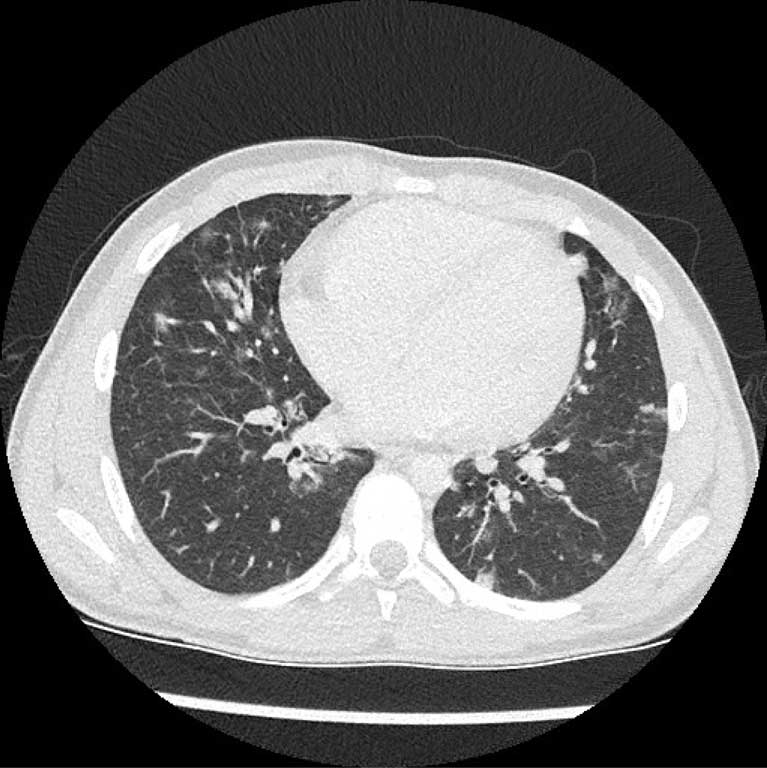
Ryc. 2. Ziarniniakowo-limfocytarne śródmiąższowe zapalenie płuc u 11-letniego chłopca z CVID, początkowo diagnozowanego w kierunku sarkoidozy. W obu płucach liczne rozsiane zmiany ogniskowe (ziarniniaki) nieostro odgraniczone oraz liczne, powiększone węzły chłonne w śródpiersiu, jako wyraz proliferacji komórek limfoidalnych w płucach i węzłach chłonnych
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Joshi AY, Iyer VN, Hagan JB et al.: Incidence and Temporal Trends of Primary Immunodeficiency: A Population-Based Cohort Study. Mayo Clin Proc 2009; 84(1): 16-22.
2. Quinti I, Soresina A, Spadaro G et al.: Long-Term Follow-Up and Outcome of a Large Cohort of Patients with Common Variable Immunodeficiency. J Clin Immunol 2007; 27(3): 308-316.
3. Bondioni MP, Duse M, Plebani A et al.: Pulmonary and Sinusal Changes in 45 Patients With Primary Immunodeficiencies. J Comput Assist Tomogr 2007; 31(4): 620-628.
4. Bonilla FA, Khan DA, Ballas ZK et al.: Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol 2015; 136(5): 1186-1205.e1-78.
5. Gathmann B, Mahlaoui N, Gèrard L et al.: Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol 2014; 134(1): 116-126.
6. Winkelstein JA, Marino MC, Lederman HM et al.: X-Linked agammaglobulinemia. Medicine 2006; 85(4): 193-202.
7. McKinney RE, Katz SL, Wilfert CM: Chronic Enteroviral Meningoencephalitis in Agammaglobulinemic Patients. Clin Infect Dis 1987; 9(2): 334-356.
8. Hampson FA, Chandra A, Screaton NJ et al.: Respiratory disease in common variable immunodeficiency and other primary immunodeficiency disorders. Clin Radiol 2012; 67(6): 587-595.
9. Pac M, Dmenska H, Bernatowska E: Pulmonary manifestation of X-linked agammaglobulinemia. Eur Respir J 2014; 44 (suppl. 58): P1239.
10. Liese JG, Wintergerst U, Tympner KD, Belohradsky BH: High- vs Low-Dose Immunoglobulin Therapy in the Long-term Treatment of X-linked Agammaglobulinemia. Arch Pediatr Adolesc Med 1992; 146(3): 335.
11. Walport MJ: Primary immunodeficiency diseases. Report of an IUIS Scientific Committee. International Union of Immunological Societies. Clin Exp Immunol 1999; suppl. 1: 1-28.
12. Wang J, Cunningham-Rundles C: Treatment and outcome of autoimmune hematologic disease in common variable immunodeficiency (CVID). J Autoimmun 2005; 25(1): 57-62.
13. Cunningham-Rundles C, Bodian C: Common Variable Immunodeficiency: Clinical and Immunological Features of 248 Patients. Clin Immunol 1999; 92(1): 34-48.
14. Urm S-H, Yun HD, Fenta YA et al.: Asthma and Risk of Selective IgA Deficiency or Common Variable Immunodeficiency: A Population-Based Case-Control Study. Mayo Clin Proc 2013; 88(8): 813-821.
15. Juhn YJ, Kita H, Yawn BP et al.: Increased risk of serious pneumococcal disease in patients with asthma. J Allergy Clin Immunol 2008; 122(4): 719-723.
16. Touw C, Ven A Van De, de Jong P: Detection of pulmonary complications in common variable immunodeficiency. Pediatr Allergy 2010; 21: 793-805.
17. Maglione PJ, Overbey JR, Radigan L et al.: Pulmonary radiologic findings in common variable immunodeficiency: clinical and immunological correlations. Ann Allergy, Asthma Immunol 2014; 113(4): 452-459.
18. Gharagozlou M, Ebrahimi FA, Farhoudi A et al.: Pulmonary complications in primary hypogammaglobulinemia: a survey by high resolution CT scan. Monaldi Arch Chest Dis 2016; 65(2): 69-74.
19. Quinti I, Soresina A, Guerra A et al.: Effectiveness of Immunoglobulin Replacement Therapy on Clinical Outcome in Patients with Primary Antibody Deficiencies: Results from a Multicenter Prospective Cohort Study. J Clin Immunol 2011; 31(3): 315-322.
20. Park JES, Beal I, Dilworth JP et al.: The HRCT appearances of granulomatous pulmonary disease in common variable immune deficiency. Eur J Radiol 2005; 54(3): 359-364.
21. Gregersen S, Aaløkken TM, Mynarek G et al.: Development of pulmonary abnormalities in patients with common variable immunodeficiency: associations with clinical and immunologic factors. Ann Allergy Asthma Immunol 2010; 104(6): 503-510.
22. Arish N, Eldor R, Fellig Y et al.: Lymphocytic interstitial pneumonia associated with common variable immunodeficiency resolved with intravenous immunoglobulins. Thorax 2006; 61(12): 1096-1097.
23. Park JH, Levinson AI: Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol 2010; 134(2): 97-103.
24. Boursiquot J-N, Gèrard L, Malphettes M et al.: Granulomatous Disease in CVID: Retrospective Analysis of Clinical Characteristics and Treatment Efficacy in a Cohort of 59 Patients. J Clin Immunol 2013; 33(1): 84-95.
25. Bates CA, Ellison MC, Lynch DA et al.: Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol 2004; 114(2): 415-421.
26. Maglione PJ, Ko HM, Beasley MB et al.: Tertiary lymphoid neogenesis is a component of pulmonary lymphoid hyperplasia in patients with common variable immunodeficiency. J Allergy Clin Immunol 2014; 133(2): 535-542.
27. Aghamohammadi A, Allahverdi A, Abolhassani H et al.: Comparison of pulmonary diseases in common variable immunodeficiency and X-linked agammaglobulinaemia. Respirology 2010; 15(2): 289-295.
28. Orange JS, Grossman WJ, Navickis RJ, Wilkes MM: Impact of trough IgG on pneumonia incidence in primary immunodeficiency: A meta-analysis of clinical studies. Clin Immunol 2010; 137(1): 21-30.
29. Cunningham-Rundles C: How I treat common variable immune deficiency. Blood 2010; 116(1): 7-15.
30. Sperlich JM, Grimbacher B, Workman S et al.: Respiratory Infections and Antibiotic Usage in Common Variable Immunodeficiency. J Allergy Clin Immunol Pract 2017. pii: S2213-2198(17)30387-2.
31. Nonas S: Pulmonary Manifestations of Primary Immunodeficiency Disorders. Immunol Allergy Clin North Am 2015; 35(4): 753-766.
32. Ozkan H, Atlihan F, Genel F et al.: IgA and/or IgG subclass deficiency in children with recurrent respiratory infections and its relationship with chronic pulmonary damage. J Invest Allergol Clin Immunol 2005; 15(151): 69-74.
33. Verma N, Grimbacher B, Hurst JR: Lung disease in primary antibody deficiency. Lancet Respir Med 2015; 3(8): 651-660.
34. Aghamohammadi A, Cheraghi T, Gharagozlou M et al.: IgA Deficiency: Correlation Between Clinical and Immunological Phenotypes. J Clin Immunol 2009; 29(1): 130-136.
35. Aghamohammadi A, Mohammadi J, Parvaneh N et al.: Progression of Selective IgA Deficiency to Common Variable Immunodeficiency. Int Arch Allergy Immunol 2008; 147(2): 87-92.
36. Juhn YJ: Risks for infection in patients with asthma (or other atopic conditions): Is asthma more than a chronic airway disease? J Allergy Clin Immunol 2014; 134(2): 247-257.e3
37. Sigal LH: Basic science for the clinician 58: IgG subclasses. J Clin Rheumatol 2012; 18(6): 316-318.
38. Nuorti JP, Whitney CG: Use of 13-Valent Pneumococcal Conjugate Vaccine and 23-Valent Pneumococcal Polysaccharide Vaccine Among Children Aged 6-18 Years with Immunocompromising Conditions: Recommendations of the Advisory Committee on Immunization Practices (ACIP). Department of Health and Human Services, Centers for Disease Control and Prevention. Weekly 2013 June 28; 62(25): 521-524.
39. Erkocoglu M, Metin A, Kaya A et al.: Allergic and autoimmune disorders in families with selective IgA deficiency. Turkish J Med Sci 2017; 47(2): 592-598.
40. Agarwal S, Cunningham-Rundles C: Assessment and clinical interpretation of reduced IgG values. Ann Allergy Asthma Immunol 2007; 99(3): 281-283.
41. Yazdani R, Azizi G, Abolhassani H, Aghamohammadi A: Selective IgA Deficiency: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Scand J Immunol 2017; 85(1): 3-12.
42. Latiff AHA, Kerr MA: The clinical significance of immunoglobulin A deficiency. Ann Clin Biochem 2007; 44(2): 131-139.
43. Kobayashi M, Bennett NM, Gierke R et al.: Intervals Between PCV13 and PPSV23 Vaccines: Recommendations of the Advisory Committee on Immunization Practices (ACIP). Morb Mortal Wkly Rep 2015; 64(34): 944-947.
44. Yel L, Ramanuja S, Gupta S: Clinical and Immunological Features in IgM Deficiency. Int Arch Allergy Immunol 2009; 150(3): 291-298.
45. Goldstein MF, Goldstein AL, Dunsky EH et al.: Pediatric selective IgM immunodeficiency. Clin Dev Immunol 2008; 2008: 624850.
46. Louis AG, Gupta S: Primary Selective IgM Deficiency: An Ignored Immunodeficiency. Clin Rev Allergy Immunol 2014; 46(2): 104-111.
47. Kung S-J, Gripp KW, Stephan MJ et al.: Selective IgM deficiency and 22q11.2 deletion syndrome. Ann Allergy Asthma Immunol 2007; 99(1): 87-92.
48. Goldstein MF, Goldstein AL, Dunsky EH et al.: Selective IgM immunodeficiency: retrospective analysis of 36 adult patients with review of the literature. Ann Allergy Asthma Immunol 2006; 97(6): 717-730.
49. Yel L, Ramanuja S, Gupta S: Clinical and immunological features in IgM deficiency. Int Arch Allergy Immunol 2009; 150(3): 291-298.
50. Ocampo CJ, Peters AT: Antibody deficiency in chronic rhinosinusitis: Epidemiology and burden of illness. Am J Rhinol Allergy 2013; 27(1): 34.
51. Fried AJ, Bonilla FA: Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections. Clin Microbiol Rev 2009; 22(3): 396-414.
52. Kim J-H, Park S, Hwang Y Il et al.: Immunoglobulin G Subclass Deficiencies in Adult Patients with Chronic Airway Diseases. J Korean Med Sci 2016; 31(10): 1560.
53. Abrahamian F, Agrawal S, Gupta S: Immunological and clinical profile of adult patients with selective immunoglobulin subclass deficiency: response to intravenous immunoglobulin therapy. Clin Exp Immunol 2010; 159(3): 344-350.
54. Popa V, Colby TV, Reich SB: Pulmonary Interstitial Disease in Ig Deficiency. Chest 2002; 122(5): 1594-1603.
55. Celińska-Löwenhoff M, Musiał J: Niedobory odporności humoralnej u osób dorosłych. Alerg Astma Immunol 2014; 19(4): 202-209.