Jacek Nikliński1, Wiesława Niklińska2, Lech Chyczewski3
Badania molekularne w rozpoznawaniu nowotworów
Molecular approaches to cancer diagnosis
1 z Kliniki Chirurgii Klatki Piersiowej Akademii Medycznej w Białymstoku
Kierownik Kliniki: dr hab. med. Jerzy Laudański
2 z Zakładu Histologii i Embriologii Akademii Medycznej w Białymstoku
Kierownik Zakładu: dr hab. Bogusław Sawicki
3 z Zakładu Klinicznej Biologii Molekularnej Akademii Medycznej w Białymstoku
Kierownik Zakładu: prof. dr hab. Lech Chyczewski
Streszczenie
Malignant tumors arise as a consequence of the accumulation of genetic and epigenetic alterations within a single cell or group of cells. A number of new molecular approaches have recently been described that may have a significant impact on cancer diagnosis and prognosis. These approaches are introduced and discussed in the context of their potential applications in clinical practice.

Badania ostatnich lat sugerują, że złośliwa zmiana nowotworowa powstaje jako wynik nagromadzenia błędów genetycznych w prawidłowej komórce, która przestaje podlegać fizjologicznym mechanizmom kontrolującym jej wzrost i różnicowanie (4). Zasadniczym zjawiskiem w przebiegu onkogenezy jest pojawienie się kolejnych mutacji aktywujących protoonkogenny i unieczynniających antyonkogeny (geny supresorowe). Końcowym efektem tego procesu jest pełne rozregulowanie aparatu genetycznego. Wieloletni proces kancerogenezy obejmuje zainicjowanie procesu przekształcania się komórek zdrowych w komórki nowotworowe, namnażanie tych komórek i nabywanie fenotypu angiogenicznego, a następnie wzrost guza pozwalający na wykrycie go badaniami obrazowymi. W tym czasie może dochodzić do powstania niezwykle istotnych dla przebiegu choroby procesów naciekania otoczenia, tworzenia mikroprzerzutów oraz nabywania przez komórki raka oporności na cytostatyki.
Szybki rozwój różnorodnych technik biologii molekularnej stał się podstawą do praktycznego zastosowania oceny zaburzeń materiału genetycznego w chorobach nowotworowych. Obecnie trwają intensywne badania nad mechanizmami predyspozycji genetycznych do chorób nowotworowych, a także poszukiwanie czynników ułatwiających wczesną diagnostykę i markerów mających znaczenie rokownicze.
Zaburzenia genetyczne pojawiające się w komórkach nowotworowych lub niejednokrotnie w zmianach przednowotworowych można analizować na poziomie DNA oraz RNA, a także produktów białkowych genów.
Badania oparte na analizie DNA
Materiałem do oceny jest DNA pochodzący z wycinków guza nowotworowego, komórek nowotworowych obecnych w płynach ustrojowych, wolnego surowiczego DNA pochodzącego z komórek nowotworowych lub leukocytów krwi obwodowej. Etapem wyjściowym dla analizy genów jest amplifikacja fragmentów badanego DNA metodą reakcji łańcuchowej z udziałem polimerazy – PCR (ang. polymerase chain reaction). Dalsze etapy mogą dostarczyć informacji o zaburzeniach w obrębie genu.
Pierwszą kategorię informacji stanowi dokładna analiza mutacji, a metodą pozwalającą na jej uzyskanie jest bezpośrednie badanie sekwencji nukleotydów w uprzednio zamplifikowanych odcinkach genu. DNA można sekwencjonować przez chemiczne rozrywanie łańcucha przy określonych zasadach (metoda Maxama-Gilberta) lub przez kontrolowane zakończenie replikacji (metoda Sangera). Ta druga metoda jest obecnie najczęściej stosowanym sposobem analizy DNA. W ostatnich latach opracowano odmianę metody Sangera, w której stosuje się znakowanie fluorescencyjnie startery (primery) lub dideoksynukleotydy. Znakowane fluorescencyjnie produkty sekwencjonowania rozdziela się w specjalnych aparatach zwanych „sekwenatorami” które automatycznie odczytują sekwencję nukleotydów w DNA i zapisują ją w pamięci komputera. Daje to możliwość błyskawicznej analizy uzyskanych danych i pozwala między innymi na wykrywanie mutacji i polimorfizmów. Metody powyższe są bardzo czułe i swoiste oraz pozwalają na dokładną lokalizację mutacji, a także na określenie jej charakteru.
Inne metody umożliwiają wykrycie mutacji bez jej pełnej charakterystyki. Do stwierdzenia obecności mutacji w określonym eksonie genu wykorzystuje się metody pośrednie, takie jak badanie polimorfizmu konformacji jednoniciowego DNA – SSCP (ang. single strand conformation polymorphism), elektroforezę w gradiencie denaturującym – DGGE (ang. denaturating gradient gel electrophoresis) oraz analizę heterodupleksów. Metoda SSCP oparta jest na zmianie konformacji (kształtu) pojedynczej nici DNA, będącej wynikiem mutacji, co prowadzi do różnicy w migracji DNA w żelu poliakrylamidowym. Metoda DGGE oparta jest na zależnej od składu zasad zdolności denaturacji DNA w różnych temperaturach i stężeniach substancji denaturującej. Analiza heterodupleksów polega na różnicy ruchliwości podczas elektroforezy na żelu poliakrylamidowym heterodupleksów (struktur dwuniciowych DNA zawierających miejsca niesparowane z powodu mutacji punktowych) w porównaniu do homodupleksów (struktur dwuniciowych w pełni komplementarnych). Metody te są łatwiejsze do wykonania w porównaniu z bezpośrednim sekwencjonowaniem genu, ale nie pozwalają na pełne scharakteryzowanie mutacji.
Opracowano ostatnio metody, które dają możliwość wykrycia mutacji występujących w bardzo małym odsetku badanej populacji komórek (2). Najczęściej taką analizę przeprowadza się stosując technikę „enriched-PCR” z jej modyfikacjami, alleloswoistej hybrydyzacji, oraz łańcuchowej reakcji ligazy (LCR). Metody te są wykorzystywane do wykrywania mutacji znanych, takich jak mutacje w kodonie 12 genu K-ras oraz mutacji w tzw. „gorących” miejscach w genie P53.
Metoda enriched-PCR polega na wybiórczej degradacji allela „dzikiego” przy zachowaniu allela zmutowanego przed amplifikacją genu w układzie PCR. W wyniku tego powstające produkty PCR stanowią jednorodną populację DNA pod względem sekwencji nukleotydów, a obecność produktu amplifikacji świadczy o obecności mutacji w badanym materiale. W przypadku, gdy oceniany na obecność mutacji kodon genu nie posiada miejsca cięcia dla żadnego ze znanych enzymów restrykcyjnych, w analizie enriched-PCR stosuje się przed etapem cięcia matrycy dodatkową amplikację DNA ze starterami określonymi jako mismatched (metodę tę określa się jako PCR-PIREMA). W wyniku tego matryca genu prawidłowego będzie przecinana przez enzym restrykcyjny, nie dając produktu amplifikacji. Natomiast w genie zmutowanym takie miejsce cięcia nie powstaje, a amplifikowany gen świadczy o obecności mutacji (ryc. 1).
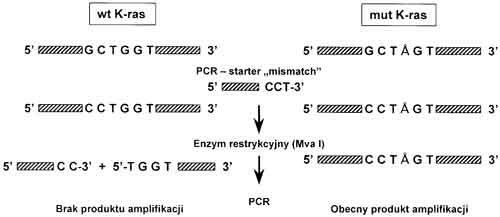
Ryc. 1. Schemat metody „PCR-PIREMA”.
Metoda alleloswoistej hybrydyzacji polega na wykorzystywaniu alleloswoistych sond molekularnych w hybrydyzacji ze zamplifikowanym genem. W tej metodzie zazwyczaj stosuje się technikę dot-blotting, w której produkty amplifikacji nanoszone są w postaci plamek na dwie błony nitrocelulozowe. Następnie jedna z błon jest hybrydyzowana z sondą o sekwencji prawidłowej, druga zaś z sondą swoistą dla sekwencji zmutowanej. Dodatni sygnał z sondą dla sekwencji zmutowanej świadczy o obecności mutacji w badanej próbce.
W metodzie LCR stosuje się powielanie in vitro fragmentu poprzez wielokrotne powtórzenie reakcji ligacji dwóch oligonukleotydów, która jest poprzedzona hybrydyzacją tych oligonukleotydów na jednej nici DNA. W metodzie tej stosowane są oligonukleotydy komplementarne do zmienionej sekwencji i powielany jest gen zmutowany (ryc. 2).
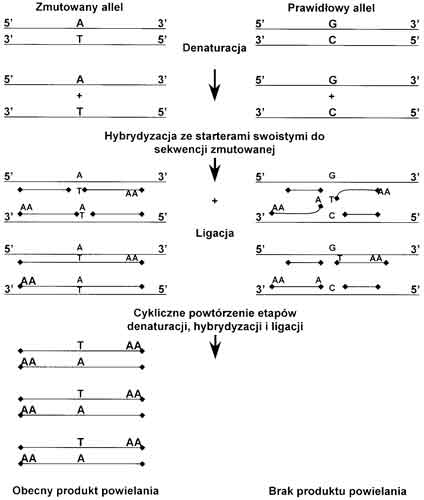
Ryc. 2. Schemat metody „LCR”.
Powyższe metody pozwalają na analizę mutacji genów, co może być wykorzystane zarówno w diagnostyce nowotworów dziedzicznych (12), jak również w ocenie molekularnej nowotworów nabytych (13). W ostatnich latach zidentyfikowano wiele genów, których mutacje predysponują do rozwoju nowotworów (18). Programy postępowania profilaktyczno-diagnostycznego zostały już opracowane dla wielu postaci nowotworów dziedzicznych. Stwarzają one nadzieję na wykrycie nowotworu we wczesnym stadium zaawansowania oraz wczesne wdrożenie postępowania terapeutycznego. W przypadku nowotworów nabytych szczególne znaczenie mają badania mutacji w celu obiektywnego wyselekcjonowania przypadków wysokiego ryzyka wystąpienia niepowodzenia leczenia. Przewidywanie przebiegu choroby u osób obciążonych nowotworem ma istotne znaczenie kliniczne, pozwala bowiem wybrać odpowiedni sposób leczenia i określić rokowanie.
W odniesieniu do raka płuca, trzustki czy jelita grubego szczególne znaczenie mają badania mutacji genów P53, P16 i K-ras (6, 9, 11). Wykazano obecność mutacji tych genów we wczesnych postaciach wyżej wymienionych nowotworów oraz w zmianach przednowotworowych. Ponadto mutacje te można wykryć izolując DNA z komórek nabłonkowych zawartych w materiale cytologicznym w stolcu, soku trzustkowym, plwocinie oraz płynie z płukania oskrzelowo-pęcherzykowego (BAL). Uzyskane wyniki wskazują, że molekularna ocena materiału cytologicznego może w przyszłości okazać się przydatną metodą wczesnego wykrywania raka płuca, a być może i innych nowotworów (1, 16).
Obecnie trwają intensywne badania nad nowymi metodami wykrywania mutacji, które pozwoliłyby na ocenę wielu genów podczas jednego badania. Taką nadzieję stwarza metoda DNA-chip, polegająca na hybrydyzacji DNA lub RNA izolowanego od chorego na nowotwór, do znacznej liczby sond reprezentujących prawidłowe i wariantowe sekwencje badanych genów umieszczonych na stałym podłożu. Analiza mutacji w tej metodzie odbywa się za pomocą skanera fluorescencyjnego wykrywającego różnice w intensywności sygnałów fluorescencji powstałych w wyniku hybrydyzacji sond do sekwencji prawidłowych i zawierających mutacje (3).
Badania z użyciem metod biologii molekularnej pozwalają nie tylko na zidentyfikowanie mutacji punktowych, lecz również na określenie ubytku fragmentów chromosomów. Oznaczanie delecji wiąże się często z badaniem utraty heterozygotyczności (LOH, ang. loss of heterozygosity). LOH jest jako utratą jednej kopii zdefiniowanego locus chromosomu, na którym zlokalizowany może być m.in. gen supresorowy. Powoduje to, że w komórce może być obecna tylko jedna kopia danego genu, którego ewentualna mutacja mogłaby doprowadzić do powstania zmienionego białka. Ostatnio najczęściej używanymi markerami genetycznymi w analizie LOH są sekwencje mikrosatelitarne. Są to wielokrotne powtórzenia sekwencji dwu- (np. CA) lub trójnukleotydowej (np. GTC). Poszczególne allele różnią się liczbą powtórzeń. W przypadku raka płuca badania cytogenetyczne i badania LOH wykazały, że często spotyka się delecje w obrębie chromosomów 3p, 5q, 8p, 9p, 13q, 17p. Wykazano tego typu zaburzenia w ok. 40% przypadków w I stadium zaawansowania niedrobnokomórkowego raka płuca (NSCLC). Ponadto zauważono, że niektóre fragmenty chromosomów ulegają delecji przede wszystkim w guzach przerzutowych (np. w niedrobnokomórkowym raku płuca są to delecje w obrębie 2q, 18q, 22q) co wskazuje, że w regionach są zlokalizowane geny hamujące inwazyjność komórek nowotworowych (7).
Innym zaburzeniem ocenianym na poziomie DNA są badania niestabilności mikrosatelitarnych. Mikrosatelity to odcinki DNA składające się z wielu powtórzeń sekwencji (np. [CA]n). W trakcie replikacji materiału genetycznego względnie łatwo dochodzi do trwałego wypętlenia nici DNA będącej matrycą lub wypętlenia nici nowo zsyntetyzowanej. Prowadzi to, odpowiednio, do wypadnięcia lub dodania w nowej nici DNA jednego lub kilku powtórzeń, z których składa się sekwencja mikrosatelitarna. W prawidłowej komórce tego typu błędy są szybko usuwane przez „system nieprawidłowo sparowanych nukleotydów” (ang. DNA mismatch repair system). Analizując próbki DNA chorych na nowotwory zauważono, że czasami pojawia się w nich różnica w długości sekwencji mikrosatelitarnych, w DNA z tkanki prawidłowej i nowotworowej pochodzących od tego samego chorego. To zjawisko jest nazywane niestabilnością sekwencji mikrosatelitarnych (ang. microsatellite instability). Wykazano częste występowanie tych zaburzeń we wrodzonym niepolipowatym raku okrężnicy. W tym typie nowotworu występuje dziedziczna mutacja jednego z genów białek systemu usuwania nieprawidłowo sparowanych nukleotydów. Niestabilność mikrosatelitarna jest również często obserwowana w innych nowotworach, np. w raku płuca i jest związana z niepomyślnym rokowaniem (2, 7).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Ahrendt S.A. et al.: Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J. Natl. Cancer. Inst. 1999, 91:332. 2. Cairns P., Sidransky D.: Molecular methods for the diagnosis of cancer. Biochim Biophys Acta 1999, 1423:C11. 3. Cheng J. et al.: Microchip-based devices for molecular diagnosis of genetic diseases. Mol. Diagn. 1997, 1:183. 4. Chorąży M.: Molekularne aspekty kancerogenezy. Nowotwory 1997, 47:251. 5. Dumont N.: Genetic and epigenetic contributions to colorectal cancer. APMIS 1999, 107:711. 6. Dziadziuszko R. et al.: Clinical implications of molecular abnormalities in lung cancer. Cancer Treat. Rev., 1998, 24:317. 7. Fong K.M. et al.: Molecular pathogenesis of lung cancer. J. Thorac. Cardiovasc. Surg. 1999, 118:1136. 8. Ghossein R.A. et al.: Molecular detection of micrometstases and circulating tumor cells in solid tumors. Clin. Cancer. Res. 1999, 5:1950. 9. Goggins M. et al.: Progress in cancer genetics: lessons from pancreatic cancer. Ann Oncol. 1999, 10 (suppl. 4):4. 10. Inga A. Et al.: Determining mutational fingerprints at the human p53 locus with a yeast functional assay: a new tool for molecular epidemiology. Oncogene 1997, 14:1307. 11. Romanowski P., Limon J.: Aberacje chromosomowe i mutacje genowe w raku jelita grubego. Nowotwory 1996, 46:213. 12. Lubiński J. i wsp.: Nowotwory dziedziczne – profilaktyka, wczesna diagnostyka i leczenie. Współczesna Onkologia 1997, 1:5. 13. Nikliński J., Furman M.: Clinical tumour markers in lung cancer. Eur. J. Cancer. Prev. 1995, 4:129. 14. Pantel K. et al.: Detection and clinical importance of micrometastatic disease. J. Natl. Cancer. Inst. 1999, 91:1113. 15. Passlick B. et al.: Detection of disseminated lung cancer cells in lymph nodes: impact on staging and prognosis. Ann. Thorac. Surg. 1996, 61:177. 16. Rimm D.L.: Molecular biology in cytopathology-current applications and future directions. Cancer Cytopathol. 2000, 90:1. 17. Salerno C.T. et al.: Detection of occult micrometastases in non-small cell lung carcinoma by reverse transcriptase-polymerase chain reaction. Chest 1998, 113:1562. 18. Steffen J.: Dziedziczne uwarunkowania w zachorowaniu na nowotwory złośliwe: udział genów o wysokiej i umiarkowanej penetracji. Nowotwory 1999, 49:71. 19. Soussi T.: p53 antibodies in the sera of patients with various types of cancer: a review. Cancer Res. 2000, 60:1777. 20. Sozzi G. et al.: Detection of microsatellite alterations in plasma DNA of non-small cell lung cancer patients: a prospect for early diagnosis. Clin. Cancer Res. 1999, 5:2689.

