© Borgis - Postępy Fitoterapii 3/2008, s. 139-164
*Tadeusz Wolski, Agnieszka Ludwiczuk, Tomasz Baj, Kazimierz Głowniak
Rodzaj Panax – systematyka, skład chemiczny, działanie i zastosowanie oraz analiza fitochemiczna nadziemnych i podziemnych organów żeń-szenia amerykańskiego – Panax quinquefolium L. Metody ekstrakcji i analizy ginsenozydów. Cz. II.
Genus panax – taxonomy, chemical composition, pharmacological effects, medicinal application and phytochemical analysis of aerial and underground parts of american ginseng (panax quinquefolium l.). method of extraction and determination of ginsenosides. part ii
Katedra i Zakład Farmakognozji z Pracownią Roślin Leczniczych, Uniwersytet Medyczny, Lublin
Kierownik Katedry i Zakładu: prof. dr hab. Kazimierz Głowniak
Summary
In this article literature review regarding extraction and identification methods of ginsenosides occurring in Panax genus was presented. Own data which concern analysis of ginsenosides fraction from Panax quinquefolium L. was also described. The occurrence of these compounds in plant material collected in different periods of ginseng vegetation as well in pharmaceutical preparations received from Panax ginseng was compared. This article contain data concerning preliminary investigations of ginseng e.g. the loss of drying and also quantitative analysis of ginsenosides by spectrophotometric method, optimization of extraction method and chromatographic separation for TLC, HPTLC, OPLC and HPLC methods. The quantitative analysis of the main compounds using TLC and HPLC methods in optimized chromatographic conditions was also performed.
Preliminary investigations concerning the loss of drying showed that underground parts of ginseng are characterized by higher content of moisture in comparison to aerial parts. The content of ginsenosides in roots as well in aerial parts of P. quinquefolium increased along with the age of the plants. The highest yield of ginsenosides was obtained in 4-year-old leaves and roots of P. quinquefolium. Ginseng leaves in comparison to ginseng roots are characterized by highest concentration of ginsenosides. Percentage of ginsenosides in roots is ranged from 6.5 to 12.5% while in ginseng leaves from 24.8 to 37.5%. Stems and fruits of Panax quinquefolium are characterized by much lower content of saponins.
Studies on isolation of ginsenosides were concerned selection of extraction solvent and extraction method. The highest extraction efficiency of ginsenosides in roots and leaves of American ginseng was observed during mechanical shaking with 50% aqueous methanol. Separation of ginsenosides conducted in 26°C and by use of chloroform – methanol – ethyl acetate – water, 15+22+40+9 (v/v) as the mobile phase was characterized by good selectivity and resolution of saponins. The best results for ginsenosides separation, using OPLC method, gave mobile phase: chloroform – methanol – ethyl acetate – water – hexane, 20+22+60+8+4 (v/v). Results from TLC, HPTLC and OPLC analyses of ginsenosides indicated that use of the different development methods resulted in significant differences between the resolutions of the compounds. Forced-flow OPLC is more sensitive method for analysis of ginsenosides.
Quantitative analysis of the main ginsenosides occurring in leaves and roots of American ginseng was executed using TLC and HPLC methods in optimized chromatographic conditions. The linear calibration curves for 6 ginsenosides, concentration ranges, limits of detection and limits of quantification for both TLC and HPLC was determined. Recovery for all investigated ginsenosides was between 88.8 and 101.2%. TLC and HPLC analyses showed that the main compound of American ginseng roots is ginsenoside Rb1. The amount of this compound was ranged from 1.36 to 3% according to extraction method and extraction solvent used. Ginsenosides Rd, Rg2 and Rb2 are the main compounds occurring in ginseng leaves. They content in plant material after mechanical shaking was 3.8, 2.4 and 1.4% respectively. The amount of another ginsenosides in ginseng leaves not exceeded 1%. Ginseng leaves, in comparison to ginseng roots are characterized by higher concentration of ginsenosides. Therefore, based on the concentration of major saponins, leaves can by alternative to root source of ginsenosides used in herbal preparations. The lack of ginsenoside Rf in aerial and undergrounds parts of Panax quinquefolium confirmed its status as a phytochemical marker differentiating American and Asian ginseng.

Niniejsza praca jest kontynuacją badań dotyczących rodzaju Panax opisanych w poprzedniej publikacji (1), w której przedstawiono ogólne wiadomości o badanym surowcu, w tym: pochodzenie i rys historyczny żeń-szenia, charakterystykę botaniczną gatunku oraz uprawę Panax quinquefolium L. Ponadto w pracy tej opisano charakterystykę ważniejszych grup związków biologicznie czynnych występujących w rodzaju Panax, tj. ginsenozydów, saponin triterpenowych, poliacetylenów, związków fenolowych oraz innych substancji biologicznie aktywnych występujących w żeń--szeniu. Ważną częścią poprzedniej pracy był opis działań leczniczych i zastosowań żeń-szenia oraz jego preparatów, ze szczególnym uwzględnieniem oddziaływania na układy: nerwowy, hormonalny oraz sercowo-naczyniowy. W tej części przedstawiono również przegląd piśmiennictwa dotyczącego działania przeciwnowotworowego, przeciwgrzybiczego i przeciwwirusowego, a także wpływ żeń-szenia i jego przetworów na układ odpornościowy, oddechowy oraz gospodarkę węglowodanową, a także działanie ochronne na wątrobę.
Najważniejszym etapem w procesie izolacji związków biologicznie czynnych występujących w surowcach roślinnych jest ich wyodrębnianie z materiału roślinnego. Dlatego też w prezentowanej publikacji przedstawiono przegląd piśmiennictwa dotyczący metod ekstrakcji oraz analizy ginsenozydów. W dalszej części prezentowanej pracy przedstawiono również wyniki własne dotyczące doboru metod ekstrakcji ginsenozydów oraz ich jakościowego i ilościowego oznaczania metodami spektroskopowymi i chromatograficznymi.
Przegląd piśmiennictwa dotyczący metod ekstrakcji ginsenozydów
Ekstrakcja ciągła metodą Soxhleta (2, 3). Jest to metoda stosunkowo tania i łatwa w wykonaniu, ponieważ nie wymaga używania specjalistycznego sprzętu; niezbędne jest jednak użycie znacznych ilości rozpuszczalników do wytrawiania substancji roślinnych i jest ona czasochłonna – to powoduje, że nie jest najchętniej stosowaną metodą ekstrakcji ginsenozydów. Badania Wu i wsp. (4) wykazały, że ekstrakcja ginsenozydów metodą Soxhleta, poprzedzająca oznaczenia ilościowe, powinna trwać od 8 do 12 godzin. Ekstrakcja tą metodą przebiega w temperaturze wrzenia rozpuszczalnika i może powodować całkowity lub częściowy rozpad termolabilnych związków. Badania niektórych autorów (5, 6) wskazują, że nie wszystkie ginsenozydy odporne są na wysoką temperaturę, szczególnie łatwo rozpadowi ulegają malonylo-ginsenozydy.
Ekstrakcja wspomagana ultradźwiękami (UAE). Surowiec traktuje się rozpuszczalnikiem i umieszcza w łaźni wodnej generującej ultradźwięki lub bezpośrednio w kolbie instaluje się sondę ultradźwiękową. Ultradźwięki wywołują zjawisko kawitacji, które powoduje znaczny wzrost ciśnienia rozpuszczalnika, co przyspiesza zarówno jego wnikanie w głąb materiału roślinnego, jak i uwalnianie substancji poprzez rozrywanie ścian komórkowych (7). Badania nad ekstrakcją ginsenozydów z żeń-szenia, prowadzone metodą ultradźwiękową, wykazały skrócenie czasu ekstrakcji z 8 do 2 godzin w porównaniu do ekstrakcji metodą Soxhleta. Ponadto do ekstrakcji wspomaganej ultradźwiękami można zastosować jako rozpuszczalnik n-butanol nasycony wodą, który okazał się lepszym ekstrahentem w porównaniu z metanolem (4).
Ekstrakcja cieczą pod zwiększonym ciśnieniem (ASE). Jest to metoda szybka (czas analizy ok. 10 min.) i niewymagająca dużych ilości rozpuszczalnika. Na przebieg i efektywność ekstrakcji mają wpływ temperatura i ciśnienie oraz rodzaj użytego ekstrahenta. Podwyższone ciśnienie w trakcie prowadzenia ekstrakcji pozwala na utrzymanie gorących rozpuszczalników w fazie ciekłej, powyżej ich temperatury wrzenia, dzięki czemu mają one korzystniejsze właściwości fizyko-chemiczne dla zapewnienia właściwego przebiegu ekstrakcji, niż w warunkach temperatury pokojowej i pod ciśnieniem atmosferycznym. Niska lepkość rozpuszczalnika w podwyższonej temperaturze powoduje lepszą penetrację badanego surowca, co z kolei zwiększa skuteczność ekstrakcji, a wyższe współczynniki dyfuzji powodują wzrost kinetyki ekstrakcji (8). W przypadku ginsenozydów najlepszym ekstrahenten metodą ASE okazał się metanol, a parametry ciśnienia i temperatury, przy których wydajność ekstrakcji jest największa, to 500 psi i 120°C. Badania Choi i wsp. (9) również wykazały wyższość metody ASE nad ekstrakcją ultradźwiękową.
Ekstrakcja wspomagana mikrofalami (MAE). W tej metodzie surowiec z rozpuszczalnikiem poddawany jest działaniu fal elektromagnetycznych o dużej częstotliwości. Zastosowany rozpuszczalnik musi być łatwo przenikany przez mikrofale, a jednocześnie powinien na tyle absorbować wydzielającą się energię, aby w krótkim czasie ekstrakcji osiągnąć wartość bliską temperatury wrzenia (10). Badania nad ekstrakcją ginsenozydów tą metodą wykazały, że efektywność ekstrakcji wspomaganej mikrofalami jest porównywalna do metody Soxhleta, jednakże czas ekstrakcji i zużycie rozpuszczalników znacznie mniejsze (czas analizy skraca się z 12 h do kilkudziesięciu sekund) (11).
Ekstrakcja przy użyciu płynu w stanie nadkrytycznym (SFE). Płyn nadkrytyczny jest ekstrahentem niepolarnym i jako taki najlepiej nadaje się do ekstrakcji związków niepolarnych. Polarność płynu można jednak w pewnych granicach zmieniać, dodając odpowiednie modyfikatory (12). Surowiec ekstrahowany jest gazem w stanie nadkrytycznym (najczęściej do tego celu używany jest dwutlenek węgla), który w odpowiednich warunkach ciśnienia i temperatury zmienia swoje właściwości fizyczne. Główną zaletą ekstrakcji płynem nadkrytycznym jest wielokrotne zwiększenie szybkości transportu masy i ciepła w porównaniu z odpowiednimi wielkościami dla cieczy, co pociąga za sobą znaczne przyspieszenie procesu ekstrakcji. Ekstrakcję ginsenozydów metodą SFE przeprowadzono stosując nadkrytyczny dwutlenek węgla w temperaturze 35-60°C, pod ciśnieniem 10,4-31,2 MPa, z dodatkiem alkoholu etylowego jako modyfikatora. Wydajność ekstrakcji była jednak niższa w porównaniu z ekstrakcją metodą Soxhleta oraz ultradźwiękami (13).
Ekstrakcja metodą Extrelut (r). Metodę tę do ekstrakcji ginsenozydów zastosowali Sticher i Soldati (14). Próbkę sproszkowanego korzenia żeń-szenia, po zmieszaniu z wodą, nanoszono na kolumnę wypełnioną granulkami nośnika. Próbka zatrzymana w porach wypełnienia tworzyła fazę stacjonarną. Przez tak przygotowaną kolumnę przepuszczano, jako eluent, n-butanol nasycony wodą. W wyniku ekstrakcji ginsenozydy przechodzą do fazy wodno-butanolowej, zaś substancje balastowe (głównie mono-, di- i oligosacharydy) są zatrzymywane na kolumnie. Odzysk ginsenozydów w tej metodzie wynosi ok. 100%.
Przegląd piśmiennictwa dotyczący metod analizy ginsenozydów
Znanych jest wiele metod analizy jakościowej i ilościowej ginsenozydów, począwszy od metod spektroskopowych, poprzez chromatografię cienkowarstwową, wysokosprawną chromatografię cieczową, kończąc na chromatografii gazowej i elektroforezie kapilarnej (15).
Metody spektroskopowe analizy ilościowej ginsenozydów opierają się na reakcji aglikonu z kwasami nieorganicznymi (np. kwasem siarkowym (VI), często w połączeniu z aldehydami aromatycznymi (waniliną, aldehydem anyżowym). Są to jednak reakcje niespecyficzne i pozwalają na oznaczenie jedynie sumy ginsenozydów (16-18).
Kolejną metodą, często stosowaną do oznaczania ginsenozydów, jest chromatografia cienkowarstwowa. Zaletą metod planarnych jest możliwość analizowania kilku próbek na jednej płytce, można też jednorazowo rozwijać kilka płytek, co umożliwia stosowanie chromatografii cienkowarstwowej jako szybkiej metody jakościowej i preparatywnej (19). W przypadku oznaczania ginsenozydów, które nie są związkami barwnymi i nie dają fluorescencji w świetle UV, ważną rolę odgrywa dobór odpowiedniego odczynnika derywatyzującego. Jego reakcja z ginsenozydami powinna być czuła, a produkty reakcji winny być trwałe. Jak wspomniano powyżej do wywoływania ginsenozydów stosuje się roztwór kwasu siarkowego (VI) i aldehydu aromatycznego (waniliny lub aldehydu anyżowego), a także pary chlorku tionylu (20-22).
W analizie ginsenozydów znalazły zastosowanie zarówno metody klasyczne: TLC (Thin-Layer Chromatography) czy HPTLC (High Performance Thin-Layer Chromatography), a także metody z wymuszonym przepływem fazy ruchomej, takie jak OPLC (Overpressured Layer Chromatography) i RPC (Rotation Planar Chromatography) (23, 24).
Najczęściej stosowaną metodą w analizie ginsenozydów jest wysokosprawna chromatografia cieczowa (HPLC – High Performance Liquid Chromatography). Głównym problemem w toku analizy HPLC ginsenozydów jest ich detekcja. Zwiazki te nie mają ugrupowań chromoforowych, które warunkują dobrą detekcję w świetle UV przy użyciu najczęściej stosowanego w HPLC detektora spektrofotometrycznego. Stwierdzono, że ginsenozydy wykazują absorbancję w dolnym zakresie widma UV (długość fali od 195 do 210 nm), dlatego też ich detekcja prowadzona jest w tym zakresie. Na wynik oznaczenia mają wpływ także składniki fazy ruchomej (wymagana jest ich wysoka czystość), ponieważ rozpuszczalniki mogą interferować z ginsenozydami (limit detekcji ginsenozydów to ok. 300 ng). Metodą alternatywną do klasycznej HPLC jest przedkolumnowa derywatyzacja, podczas której do związków dołączane są grupy chromoforowe, pozwalające na przeprowadzenie analizy przy wyższych długościach fali (np. 254 nm). Lepsze wyniki analizy HPLC ginsenozydów można osiągnąć stosując zamiast detektora spektrofotometrycznego inne detektory, np. detektor fluorescencyjny, detektor aerozolowy promieniowania rozproszonego (ELSD), czy też detektor masowy (MS lub MS/MS). Zastosowanie chromatografii cieczowej w połączeniu ze spektrometrią masową (LC-MS) obniża limit detekcji ginsenozydów z 300 ng do 2 pg (25-27).
Metodą rzadziej stosowaną przy ilościowym oznaczaniu ginsenozydów jest chromatografia gazowa (GC). Metoda ta ma wysoką czułość, ale ginsenozydy, jako związki nielotne, przed wprowadzeniem na kolumnę, muszą być zhydrolizowane i poddane trimetylosilanizacji. W konsekwencji tą metodą można oznaczać tylko sapogeniny: protopanaxadiol, protopanaxatriol i kwas oleanolowy. Chromatografia gazowa ma więc zastosowanie wówczas, gdy dla oceny surowca, czy ekstraktu, wystarcza ogólna zawartość sumy ginsenozydów (28, 29).
Przegląd piśmiennictwa dotyczącego analizy ginsenozydów metodami chromatograficznymi przedstawia tabela 1.
Tabela 1. Przegląd metod chromatograficznych stosowanych do oznaczania ginsenozydów (49).
| Metoda | Płytka/Kolumna | Faza ruchoma | Detekcja | Piśmiennictwo |
| TLC | Żel krzemionkowy F254
Żel krzemionkowy 60 | CHCl3-MeOH-H2O (6:4:1)
n-BuOH-EtOAc-H2O (4:1:5) (warstwa górna) | 1% Ce(SO4)/10% H2SO4 odczynnik Libermanna-Burcharda | (30)
(31) |
Żel krzemionkowy 100 F254
Żel krzemionkowy 60 | CHCl3-MeOH-H2O (14:8:1)
CHCl3-EtOAc-MeOH-H2O (15:50:22:10)
oraz CHCl3-MeOH-H2O (69:27:4) | wanilina w H2SO4
wanilina w H2SO4 | (32)
(33) |
| Żel krzemionkowy G | CHCl3-MeOH-H2O (65:50:10) | odczynnik Godina (1% wanilina/5% H2SO4) | (34) |
| HPTLC | Żel krzemionkowy F254 | CHCl3-EtOAc-MeOH-H2O (15:40:22:9) | aldehyd anyżowy | (20) |
| Żel krzemionkowy G60 | 1,2-dichloroetan-EtOH-MeOH-H2O (56,8:19,2:19,2:4,8) | pary chlorku tionylu | (21) |
| OPLC | Żel krzemionkowy G60 | metylo-etylo-keton-MeOH-H2O (80:10:10) | wanilina w H2SO4 | (35) |
| RPC | Żel krzemionkowy | metylo-etylo-keton-MeOH-H2O-heksan (70:22:8:30)
metylo-etylo-keton-MeOH-H2O-heksan (80:10:10:15) | wanilina w H2SO4 | (22) |
| HPLC | μPorasil | n-heptan-n-BuOH-ACN-H2O (1000:446:132:36) | λ=207 nm | (14) |
μBondapack C18
μBondapack C18 | MeOH-H2O (440:560)
ACN-H2O (30:70) i ACN-H2O (18:82) | λ=207 nm
λ=203 nm | (14)
(36) |
| Resolve C18 | bufor fosforanowy (pH=8,5-8,1) +ACN+H2O (gradient) | λ=203 nm | (37) |
| Mightysil RP-18 | ACN-H2O-5%CH3COOH (15:80:5) i ACN-H2O (80:20) (gradient) | ELSD | (38) |
| LiChrosorb NH2 | 0,3 mM 2-t-butyloantrachinon w ACN-H2O (4:1) | detektor fluorescencyjny | (39) |
| Superspher 100 RP-18e | ACN-H2O-0,1%H3PO4 (21:72:8) i ACN (gradient) | λ=202 nm | (40) |
| LiChrosorb NH2 | MeOH-ACN-etanodiol-0,14M NH4OAc (150:350:25:53) | detektor refraktometryczny | (41) |
| LiChrosorb NH2 | H2O-2-propanol-ACN (gradient) | ELSD | (42) |
| Atlantis C18 | mrówczan amonu-ACN i ACN (gradient) | MS/MS | (43) |
| Nova-Pak C18 | 50% aq. MeOH z 0.1% THF i 90% aq. MeOH (gradient) | detektor fluorescencyjny | (27) |
| Symetry C18 | ACN-H2O (gradient) | λ=203 nm | (44) |
| Phenomenex Luna C18 | 10 mM NH4OAc-ACN | MS | (44) |
| GC | SE-30 Chromasorb W AW DMCS | hel | FID | (45) |
| Se-30 Chromasorb G | azot | FID | (31) |
| DB-5 oraz HP-1 | hel | MS | (28) |
| HP-1 | hel | MS | (29) |
Jak podano we wstępie, celem prezentowanej pracy jest przegląd piśmiennictwa dotyczący metod ekstrakcji ginsenozydów z rodzaju Panax oraz ich analiza metodami spektroskopowymi i chromatograficznymi. Celem naszych badań był dobór optymalnych warunków ekstrakcji oraz porównanie metod jakościowego i ilościowego oznaczania ginsenozydów, głównie metodami chromatograficznymi TLC i HPLC.
Materiał i metody
Materiał do badań stanowiły organy podziemne (korzenie) i nadziemne (liście, łodygi, owoce) żeń- -szenia pięciolistnego ( Panax quinquefolium L.) uprawianego w Katedrze Roślin Leczniczych i Przemysłowych Uniwersytetu Przyrodniczego w Lublinie oraz na plantacji żeń-szenia w Suchodołach koło Fajsławic.
W celach porównawczych analizie poddano również preparat galenowy, będący alkoholowym, gęstym wyciągiem z Panax ginseng ( Extractum ginseng spir. spissum) oraz preparat farmaceutyczny Panaxan(r). Powyższe formy preparatów były wyprodukowane przez Phytopharm, Klęka.
W analizie stosowano wzorce ginsenozydów: Rb1, Rb2, Rc, Re, Rd, Rf, Rg1 produkcji Roth, Niemcy.
Oznaczenie straty masy po suszeniu
Oznaczenie wykonano metodą wagową zgodnie z FP V (46) i FP VI (47).
Oznaczanie sumy ginsenozydów
Oznaczenie zawartości procentowej sumy ginsenozydów prowadzono metodą spektrofotometryczną opisaną w piśmiennictwie (18, 48). Zawartość procentową ginsenozydów podano w przeliczeniu na ginsenozyd Rg1. Analizie poddano organy nadziemne i podziemne Panax quinquefolium w I, II, III i IV roku wegetacji. Do wykonania oznaczeń odważono po około 0,15 g korzeni, 0,1 g liści, 0,5 g łodyg i 0,3 g owoców.
Celem porównania zawartości sumy ginsenozydów w preparatcie galenowym Extractum ginseng spir. spissum odważono 0,208 g alkoholowego, gęstego wyciągu z żeń-szenia właściwego, zaś do analizy preparatu Panaxan użyto 1 kapsułkę o masie 0,308 g.
Wyodrębnianie frakcji ginsenozydów
Ze względu na najwyższą zawartość ginsenozydów
w surowcach 4-letnich, badaniom poddano 4-letnie korzenie i liście żeń-szenia pięciolistnego ( Panax quinquefolium L.). W celu wyizolowania frakcji ginsenozydów z badanych surowców zastosowano następujące metody ekstrakcyjne: ekstrakcję przez wytrząsanie, ekstrakcję wspomaganą ultradźwiękami (UAE) oraz ekstrakcję cieczą pod zwiększonym ciśnieniem (ASE) (49). Do ekstrakcji odważano każdorazowo po 2 g surowca.
Ekstrakcja przez wytrząsanie. Surowiec umieszczano w kolbie okrągłodennej o pojemności 250 ml i dodawano 50 ml rozpuszczalnika. Jako ekstrahentów użyto: 50, 75 oraz 100% metanolu, a także 50 i 100% etanolu i n-propanolu. Zawartość kolby wytrząsano na wytrząsarce przez 15 min. Następnie rozpuszczalnik zdekantowano, a surowiec traktowano nową porcją ekstrahenta. Ekstrakcję powtarzano 3-krotnie. Tak otrzymany ekstrakt odparowano do sucha na wyparce obrotowej w temperaturze 60°C. Suchą pozostałość rozpuszczano w 50% MeOH i przenoszono ilościowo do kolbek miarowych o pojemności 25 ml.
Ekstrakcja wspomagana ultradźwiękami (UAE). Surowiec umieszczano w kolbie okrągłodennej o pojemności 250 ml i dodawano 50 ml rozpuszczalnika. Jako ekstrahentów używano: czystego metanolu oraz wodnych roztworów metanolu o stężeniu 50 i 75%. Kolby z zawartością umieszczano w łaźni generującej ultradźwięki, pod chłodnicą zwrotną i ekstrahowano 15 min. w temperaturze 60 oraz 80°C. Ekstrakcję powtarzano 3-krotnie. Otrzymane ekstrakty odparowywano do sucha na wyparce obrotowej w temp. 60°C. Suchą pozostałość rozpuszczano w 50% MeOH i przenoszono ilościowo do kolbek miarowych o pojemności 25 ml.
Ekstrakcja cieczą pod zwiększonym ciśnieniem (ASE). Odważone próbki surowca umieszczano w stalowej gilzie aparatu do ekstrakcji ciśnieniowej ASE 100 firmy Dionex. Surowiec ekstrahowano metanolem (50, 75 i 100%) w temperaturze 40, 60, 80, 100 i 120°C. Próbki ekstrahowano 3-krotnie, a całkowity czas ekstrakcji wynosił około 25 min. Otrzymane ekstrakty odparowywano do sucha na wyparce obrotowej w temp. 60°C. Suchą pozostałość rozpuszczano w 50% MeOH i przenoszono ilościowo do kolbek miarowych o pojemności 25 ml.
Otrzymane w opisany powyżej sposób ekstrakty poddano oczyszczaniu metodą ekstrakcji do fazy stałej (SPE). Tok postępowania obejmował oczyszczanie wyciągów metanolowo-wodnych (50%) z substancji balastowych oraz elucję ginsenozydów z sorbentu z chemicznie związaną fazą oktadecylową (C18, 500 mg).
Etapy procesu SPE (49):
1. Kondycjonowanie sorbentu C18
Przez kolumienki zamontowane na komorze do
sączenia próżniowego przesączono kolejno:
– 10 ml metanolu,
– 10 ml wody, pozostawiając 0,5 cm warstwę cieczy nad złożem.
2. Dozowanie wyciągów
Przez kondycjonowane złoże przesączono do od-bieralników pod próżnią (0,02 MPa) 10 ml wyciągów z liści i korzeni żeń-szenia
3. Przemywanie złoża (elucja balastów)
Po przesączeniu wyciągów z żeń-szenia złoże przemyto kolejno:
– 10 ml wody,
– 10 ml 30% metanolu.
4. Elucja ginsenozydów z sorbentu C18
Ginsenozydy zaadsorbowane na kolumienkach wymywano 10 ml czystego metanolu i przenoszono ilościowo do kolbek miarowych o pojemności 10 ml.
Ocena składu jakościowego i ilościowego ginsenozydów
Chromatografia cienkowarstwowa
Analizie chromatograficznej poddano frakcje ginsenozydów wyizolowane z części nadziemnych i podziemnych żeń-szenia pięciolistnego zgodnie z wcześniej opisanymi procedurami. Jednocześnie przygotowano roztwory wzorcowe ginsenozydów Rb1, Rb2, Rg1, Rc, Rd, Re i Rf o stężeniu 0,5 mg/ml. Celem porównania składu frakcji ginsenozydów występujących w Panax quinquefolium i P. ginseng analizie poddano również alkoholowy, gęsty wyciąg Extractum ginseng spir. spissum i preparat Panaxan(r). Do analizy użyto 1 kapsułkę preparatu Panaxan(r), oraz 0,15 g alkoholowego, gęstego wyciągu z żeń-szenia właściwego. Ginsenozydy ekstrahowano 50% metanolem zgodnie z wcześniej opisanymi procedurami (49).
Analizę cienkowarstwową prowadzono na płytkach szklanych lub foliach aluminiowych (TLC i HPTLC) pokrytych żelem krzemionkowym G i GF254. Płytki rozwijano w komorach pionowych, płaskich typu DS oraz w płaskiej komorze termostatowanej DST. Chromatogramy wywoływano przez spryskiwanie odczynnikiem Godina (5% roztwór H2SO4 w etanolu i 1% roztwór waniliny w etanolu) i ogrzewano w temperaturze 105°C przez 10 min. Następnie wykonano zdjęcia otrzymanych chromatogramów w świetle widzialnym przy użyciu urządzenia Videoscaner TLC/HPTLC (Camag, Szwajcaria) i poddano obróbce densytometrycznej przy użyciu densytometru firmy Camag (Szwajcaria) oraz J&M (Niemcy) przy dłu-
gości fali λ=540 nm. (49).
Pierwszym etapem analizy cienkowarstwowej ginsenozydów była optymalizacja warunków rozdziału chromatograficznego, tj. a – dobór odpowiedniej fazy ruchomej i b – dobór optymalnej temperatury rozdzielania.
a) Dobór fazy ruchomej
Do badań wykorzystano następujące fazy ruchome:
(1) keton metylowo-etylowy – metanol – woda,
80+10+10 ( v/v) (35),
(2) keton metylowo-etylowy – metanol – woda –
heksan, 70+22+8+30 ( v/v) (22),
(3) keton metylowo-etylowy – metanol – woda –
heksan, 80+10+10+15 ( v/v) (22),
(4) chloroform – metanol – woda, 65+50+10 ( v/v)
(48, 34),
(5) chloroform – octan etylu – metanol – woda,
15+40+22+9 ( v/v) (20, 50),
(6) chloroform – octan etylu – metanol – woda –
heksan, 20+60+22+8+4 ( v/v) (23, 49, 50),
(7) chloroform – octan metylu – metanol – woda –
heksan, 20+60+22+8+4 ( v/v) (23, 49),
(8) chloroform – octan propylu – metanol – woda–
heksan, 20+60+22+8+4 ( v/v) (23, 49)
(9) chloroform – octan butylu – metanol – woda –
heksan, 20+60+22+8+4 ( v/v) (23, 49).
b) Dobór optymalnej temperatury rozwijania
Analizę cienkowarstwową ginsenozydów (mieszanina 7 wzorcowych substancji) prowadzono w płaskiej komorze termostatowanej DST przy użyciu dwóch faz ruchomych: (5) chloroform – octan etylu – metanol – woda, 15+40+22+9 ( v/v) oraz (6) chloroform – octan etylu – metanol – woda – heksan, 20+60+22+8+4 ( v/v). Płytki rozwijano w następujących temperaturach: 17, 22, 24, 26, 28 i 30°C.
Analiza jakościowa ginsenozydów oraz cukrów i aglikonów uwolnionych po hydrolizie kwaśnej
Analizę jakościową wyodrębnionych frakcji ginsenozydów wykonano poprzez porównanie wartości Rf pasm obecnych na chromatogramach z wartościami Rf wzorcowych substancji. Płytki rozwijano przy użyciu dwóch faz ruchomych: (5) chloroform – octan etylu – metanol – woda, 15+40+22+9 ( v/v) oraz (6) chloroform – octan etylu – metanol – woda – heksan, 20+60+22+8+4 ( v/v).
Drugim etapem analizy jakościowej ginsenozydów była hydroliza saponin i analiza TLC cukrów i aglikonów uwolnionych po hydrolizie.
Hydroliza kwaśna: Hydrolizę wykonano zgodnie z metodyką opisaną przez Ouyanga (51). Ekstrakt z żeń-szenia otrzymany przez wytrząsanie z 50% metanolem, zgodnie z procedurą opisaną wcześniej, odparowywano do sucha pod ciśnieniem 1-1,5 kPa. Suchą pozostałość rozpuszczano w 20 ml 5% roztworu kwasu siarkowego (VI) w 50% MeOH i ogrzewano na łaźni wodnej pod chłodnicą zwrotną przez 3 godziny. Po tym czasie kolbę z hydrolizatem studzono, rozcieńczano dwukrotnie wodą i zobojętniano 2% roztworem wodorotlenku sodu.
Roztwory wodne otrzymane w wyniku hydrolizy ekstrahowano następnie chloroformem (5x20 ml). W ten sposób otrzymano warstwę organiczną (chloroformową) zawierającą aglikony (sapogeniny) oraz warstwę wodną zawierającą cukry. Oba roztwory odparowano do sucha na wyparce próżniowej, a suche pozostałości rozpuszczano w niewielkiej ilości metanolu i poddawano analizie TLC. Badania na obecność aglikonów prowadzono na płytkach żelowych w dwóch układach:
– chloroform – metanol, 10+1 ( v/v) (52)
– benzen – octan etylu, 1+1 ( v/v) (53).
Otrzymane chromatogramy wywoływano przez spryskanie odczynnikiem Godina.
Badania na obecność cukrów prowadzono na płytkach szklanych pokrytych celulozą w komorach stojących. Płytki rozwijano dwukrotnie na stałym dystansie przy użyciu układu rozwijającego: n-butanol – pirydyna – woda, 6+4+3 ( v/v) (54, 55). Rozwinięte chromatogramy spryskiwano ftalanem aniliny i ogrzewano w temperaturze 105°C przez 5 min. Analizę jakościową wyodrębnionych frakcji ginsenozydów wykonano poprzez porównanie wartości Rf pasm obecnych na chromatogramach z wartościami Rf wzorcowych substancji.
Analiza jakościowa ginsenozydów metodą OPLC
Analizę ginsenozydów metodą OPLC prowadzono przy użyciu aparatu firmy OPLC-NIT (Węgry) na foliach aluminiowych HPTLC (20x20 cm) i płytkach szklanych HPTLC (10x20) pokrytych żelem krzemionkowym Si 60G F254. Do badań użyto trzech układów rozwijających: (5) chloroform – octan etylu – metanol – woda, 15+40+22+9 ( v/v); (6) chloroform – octan etylu – metanol – woda – heksan, 20+60+22+8+4 ( v/v) oraz (8) chloroform – octan propylu – metanol – woda – heksan, 20+60+22+8+4 ( v/v).
Zastosowano dwie techniki rozwijania: liniową i kołową.
Rozwijanie liniowe prowadzono pod ciśnieniem 40 barów w czasie 50 min. Przepływ fazy ruchomej wynosił 50 μl/min dla płytek o wymiarach 10x20 cm oraz 80 μl/min dla płytek 20x20 cm; objętość startowa fazy ruchomej – 150 μl (płytka 10x20 cm) i 300 μl (płytka 20x20); objętość fazy ruchomej użyta do analizy – 2500 μl (płytka 10x20 cm) i 4000 μl (płytka 20x20).
Rozwijanie kołowe prowadzono na płytkach o wymiarach 20x20 cm pod ciśnieniem 40 barów w czasie 9 min. Przepływ fazy ruchomej wynosił 450 μl/min; zużyto 4000 μl fazy ruchomej.
„Pre-run” dla obu technik rozwijania prowadzono pod ciśnieniem 40 barów przez 10 min. Jako fazy ruchomej użyto octanu etylu, przy szybkości przepływu 1500 μl/min.
Analiza ilościowa ginsenozydów metodą densytometryczną
Analizę ilościową ginsenozydów prowadzono na płytkach szklanych w komorze termostatowanej w temperaturze 26°C przy użyciu fazy ruchomej (5) chloroform – octan etylu – metanol – woda, 15+40+22+9 ( v/v). W zależności od stężenia ginsenozydów we frakcjach wyodrębnionych z liści i korzeni żeń-szenia pięciolistnego, na płytki nanoszono po 5, 10 lub 20 μl wyciągu w trzech powtórzeniach. Jednocześnie wykonywano krzywe wzorcowe ginsenozydów nanosząc na płytki po 1, 2, 5, 10 i 15 μl wzorców o stężeniu 0,5 mg/ml. Tak otrzymane płytki wywoływano odczynnikiem Godina i poddano obróbce densytometrycznej przy długości fali λ=540 nm.
Analiza jakościowa i ilościowa frakcji ginsenozydów metodą HPLC
Analizie HPLC poddano frakcje ginsenozydów wyizolowane z części nadziemnych i podziemnych żeń-szenia pięciolistnego zgodnie z procedurą opisaną wcześniej.
Analizę jakościową i ilościową HPLC prowadzono w oparciu o nowo opracowaną procedurę wykorzystującą zoptymalizowane warunki rozdzielania. Badania prowadzono przy użyciu chromatografu cieczowego LaChrom firmy Merck wyposażonego w zawór dozujący typu Rheodyne, o objętości pętli dozownika równej 20 μl, detektor z matrycą diodową (DAD) oraz termostat i degazer. Rozdział ginsenozydów prowadzono w temperaturze 23°C na kolumnie LiChrocart (250x4,6 mm, dp=5 μm). Zastosowano gradientową metodę rozdzielania przy zastosowaniu dwuskładnikowej fazy ruchomej: acetonitryl i woda (tab. 2) oraz detekcję związków przy długości fali λ=195 nm.
Tabela 2. Profil gradientu zastosowany w analizie HPLC ginsenozydów (49).
| Czas (min) | ACN (%) | H2O (%) | Przepływ (ml/min) |
0
20
25
30
35
45
47
50
52 | 18
18
30
30
35
40
45
18
18 | 82
82
70
70
65
60
55
82
82 | 1,5
1,5
1,0
1,0
1,0
1,0
1,0
1,5
1,5 |
Identyfikację związków prowadzono porównując czasy retencji pików związków obecnych na chromatogramach z czasami retencji pików substancji wzorcowych oraz metodą dodatku wzorca wewnętrznego. Analizę ilościową ginsenozydów prowadzono metodą krzywej wzorcowej. W tym celu przygotowano wzorce o stężeniach 0,5 mg/ml, z których do krzywej kalibracyjnej sporządzono dodatkowo cztery poziomy rozcieńczeń: 0,025; 0,05; 0,1 i 0,25 mg/ml. Odniesienie pól powierzchni pików ginsenozydów (zawartych w analizowanych próbkach), mających czasy retencji odpowiadające substancjom wzorcowym, do odpowiednich krzywych wzorcowych, umożliwiło obliczenie zawartości tych związków w poszczególnych frakcjach.
WYNIKI I OMÓWIENIE
Strata masy po suszeniu badanych surowców
Dane dotyczące straty masy po suszeniu w organach
podziemnych i nadziemnych żeń-szenia pięciolistnego w I, II, III, IV roku wegetacji przedstawia tabela 3.
Tabela 3. Strata masy po suszeniu w różnych organach Panax quinquefolium L. (SD dla n=3) (24).
| Rok wegetacji | Zawartość wilgoci (%) |
| korzeń | liść | łodyga | owoc |
| I | 7,79 ? 0,01 | 6,89 ? 0,02 | 6,87 ? 0,11 | - |
| II | 8,23 ? 0,05 | 7,92 ? 0,06 | 7,57 ? 0,09 | 6,14 ? 0,01 |
| III | 7,89 ? 0,03 | 7,39 ? 0,01 | 6,69 ? 0,08 | 5,78 ? 0,01 |
| IV | 8,57 ? 0,03 | 7,47 ? 0,06 | 6,86 ? 0,05 | 6,03 ? 0,02 |
Z danych przedstawionych w tabeli 3 wynika, że najwyższą stratą masy po suszeniu charakteryzują się korzenie, dla których waha się ona w granicach 7,79% (korzeń 1-roczny) do 8,57% (korzeń 4-letni). W przypadku części nadziemnych roślin strata masy po suszeniu mieściła się w granicach:
liście: od 6,89% (1-roczne) do 7,92% (2-letnie),
łodygi: od 6,69% (3-letnie) do 7,57% (2-letnie),
owoce: od 5,78% (3-letnie) do 6,14% (2-letnie).
Najwyższą stratą masy po suszeniu organów nadziemnych charakteryzują się liście, łodygi i owoce w drugim roku wegetacji. Z tabeli 3 wynika również, że zmianę straty masy po suszeniu w poszczególnych organach żeń-szenia pięciolistnego w każdym roku wegetacji można przedstawić następująco: korzenie> liście ≥ łodygi > owoce
Graficzny obraz, zmiany straty masy po suszeniu w poszczególnych organach żeń-szenia w I, II, III i IV roku wegetacji, przedstawia rycina 1.
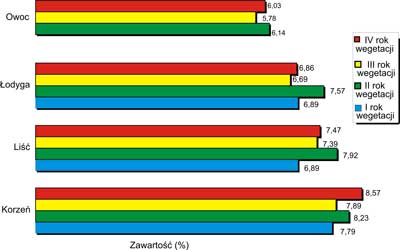
Ryc. 1. Strata masy po suszeniu w poszczególnych organach Panax quinquefolium w I, II, III i IV roku wegetacji (49).
Wyniki oznaczeń zawartości sumy ginsenozydów
Dane liczbowe dotyczące zawartości sumy ginsenozydów w przeliczeniu na ginsenozyd Rg1 w organach nadziemnych i podziemnych Panax quinquefolium w I, II, III i IV roku wegetacji przedstawia tabela 4.
Tabela 4. Zawartość sumy ginsenozydów w przeliczeniu na ginsenozyd Rg1 w różnych organach żeń-szenia amerykańskiego. (SD dla n=4) (24, 49).
| Rok wegetacji | Zawartość ginsenozydów (%) |
| korzeń | liść | łodyga | owoc |
| I | 6,550 ? 0,468 | 33,750 ? 1,183 | 5,150 ? 0,145 | - |
| II | 8,552 ? 0,465 | 25,498 ? 1,211 | 4,785 ? 0,071 | 3,685 ? 0,406 |
| III | 7,325 ? 0,212 | 24,815 ? 0,524 | 3,978 ? 0,276 | 3,288 ? 0,191 |
| IV | 12,530 ? 0,063 | 37,505 ? 0,361 | 5,620 ? 0,192 | 3,385 ? 0,384 |
Jak wynika z tabeli 4 procentowa zawartość sumy ginsenozydów w przeliczeniu na Rg1 w korzeniach żeń-szenia pięciolistnego waha się w granicach 6,5% dla korzenia 1-rocznego do 12,5% dla korzenia 4-letniego. Stanowi to prawie 2-krotny wzrost zawartości ginsenozydów w trakcie rozwoju rośliny. Interesująco przedstawia się zawartość ginsenozydów w częściach nadziemnych Panax quinquefolium L. Podczas gdy w łodygach i owocach waha się ona w granicach od około 3,3 do 5,6%, to w liściach zawartość ginsenozydów jest kilkakrotnie wyższa i mieści się w granicach od 24,8 do 37,5%. Różnice w zawartości ginsenozydów w poszczególnych organach w różnym czasie rozwoju rośliny dobrze obrazuje rycina 2.
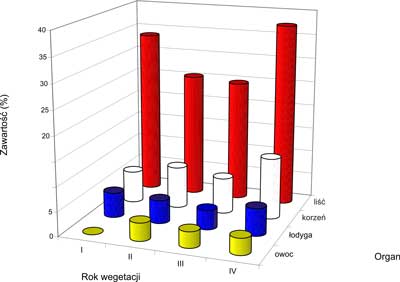
Ryc. 2. Zawartość ginsenozydów w różnych organach żeń-szenia w czasie rozwoju rośliny (49).
Jak wynika z ryciny 2 zmiany zawartości ginsenozydów w ontogenezie żeń-szenia pięciolistnego dla poszczególnych organów można przedstawić następująco:
liście: (I> II> III)
korzenie: (I
III)
łodygi: (I> II> III)
owoce: II> III IV
gdzie: I – pierwszy rok uprawy; II – drugi rok uprawy; III – trzeci rok uprawy; IV – czwarty rok uprawy.
Z danych tych wynika, że wszystkie organy w IV roku uprawy żeń-szenia pięciolistnego, za wyjątkiem owoców, charakteryzują się największą zawartością ginsenozydów.
W dalszej części pracy wyznaczono zawartość sumy ginsenozydów również w preparatach produkowanych na bazie korzeni Panax ginseng, tj. kapsułkach Panaxan(r), których zawartość stanowi Ginseng radix w ilości 30 mg/kapsułkę oraz Extractum ginseng spir. spissum, który stanowi półprodukt do preparatów farmaceutycznych. Dane dotyczące zawartości ginsenozydów w tych preparatach przedstawia tabela 5.
Tabela 5. Zawartość sumy ginsenozydów w przeliczeniu na ginsenozyd Rg1 w preparatach z żeń-szenia właściwego. (SD dla n=4).
| Preparat | Oznaczona zawartość ginsenozydów (%) | Zawartość ginsenozydów wymagana przez producenta |
| Extractum ginseng spir. spissum | 17,92 ? 0,79 | nie mniej niż 12% |
| Panaxan(r) | 4,99 ? 0,30
(14,98 ? 0,90 mg/kapsułkę) | od 3% do 5%
(min. 9 mg/kapsułkę) |
Jak wynika z tabeli 5 zawartość ginsenozydów w Extractum ginseng spir. spissum wynosi średnio 17,92% a w preparacie Panaxan(r) – 4,99%, co stanowi 14,98 mg/kapsułkę. Z danych tych wynika, że badane preparaty spełniają wymagania ich producentów, co do zawartości ginsenozydów.
Wyniki analizy jakościowej ginsenozydów metodami chromatograficznymi
Poniżej przedstawiono wyniki optymalizacji rozdziału ginsenozydów metodami TLC, HPTLC, OPLC oraz HPLC.
Wyniki optymalizacji rozdziału ginsenozydów metodą chromatografii cienkowarstwowej (TLC)
Pierwszym etapem analizy cienkowarstwowej ginsenozydów występujących w częściach nadziemnych i podziemnych żeń-szenia pięciolistnego był dobór odpowiedniej fazy ruchomej. Spośród 9 testowanych faz ruchomych, opisanych wcześniej, do analizy TLC najlepsze okazały się:
(5) chloroform – octan etylu – metanol – woda, 15+40+22+9 ( v/v) (20, 50, 56, 57),
(6) chloroform – octan etylu – metanol – woda – heksan, 20+60+22+8+4 ( v/v) (50, 56, 58)
Kolejnym etapem doboru optymalnych warunków rozdziału chromatograficznego był dobór temperatury rozdzielania. Badania prowadzono w termostatowanej komorze typu sandwicz przy użyciu dwóch wcześniej wybranych faz ruchomych w następujących temperaturach: 17, 22, 24, 26, 28 i 30°C. Wpływ temperatury na wartości współczynników Rf ginsenozydów w następujących fazach ruchomych: (5) oraz (6) przedstawia rycina 3. Wartości współczynników Rf i Rs dla 7 wzorcowych ginsenozydów rozwijanych w dwu testowanych układach przedstawiają tabele 6 i 7.
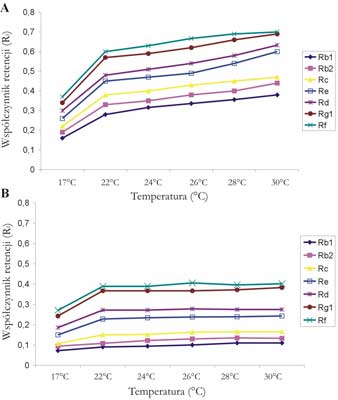
Ryc. 3. Wpływ temperatury rozdzielania na wartości współczynników Rf ginsenozydów w obu badanych fazach ruchomych. A – chloroform – octan etylu – metanol – woda, 15+40+22+9 (v/v). B – chloroform – octan etylu – metanol – woda – heksan, 20+60+22+8+4 (v/v).
Tabela 6. Wartości współczynników Rf i Rs dla mieszaniny wzorcowych ginsenozydów poddanych analizie TLC w różnych warunkach temperatury; faza ruchoma (5): chloroform – metanol – octan etylu – woda, 15+22+40+9 (v/v).
| Związek | Temperatura |
| 17°C | 22°C | 24°C | 26°C | 28°C | 30°C |
| Rf | Rs | Rf | Rs | Rf | Rs | Rf | Rs | Rf | Rs | Rf | Rs |
| Rb1 | 0.16 | | 0.28 | | 0.32 | | 0.34 | | 0.36 | | 0.38 | |
| 1.2 | 1.7 | 1.4 | 1.8 | 1.4 | 2.1 |
| Rb2 | 0.19 | | 0.33 | | 0.35 | | 0.38 | | 0.40 | | 0.44 | |
| 1.2 | 2.0 | 1.8 | 2.0 | 1.8 | 1.1 |
| Rc | 0.22 | | 0.38 | | 0.40 | | 0.43 | | 0.45 | | 0.47 | |
| 1.8 | 3.0 | 2.7 | 2.6 | 3.2 | 4.6 |
| Re | 0.26 | | 0.45 | | 0.47 | | 0.49 | | 0.54 | | 0.60 | |
| 1.6 | 1.3 | 1.4 | 2.0 | 1.4 | 1.1 |
| Rd | 0.30 | | 0.48 | | 0.51 | | 0.54 | | 0.58 | | 0.63 | |
| 1.8 | 1.2 | 3.5 | 3.4 | 2.8 | 2.1 |
| Rg1 | 0.34 | | 0.57 | | 0.59 | | 0.62 | | 0.66 | | 0.69 | |
| 1.2 | 1.8 | 1.4 | 2.0 | 1.1 | 0.7 |
| Rf | 0.37 | | 0.60 | | 0.63 | | 0.67 | | 0.69 | | 0.70 | |
| | | | | |
Tabela 7. Wartości współczynników Rf i Rs dla mieszaniny wzorcowych ginsenozydów poddanych analizie TLC w różnych warunkach temperatury; faza ruchoma (6): chloroform – metanol – octan etylu – woda– heksan, 20+22+60+8+4 (v/v).
| Związek | Temperatura |
| 17°C | 22°C | 24°C | 26°C | 28°C | 30°C |
| Rf | Rs | Rf | Rs | Rf | Rs | Rf | Rs | Rf | Rs | Rf | Rs |
| Rb1 | 0.07 | | 0.09 | | 0.09 | | 0.10 | | 0.11 | | 0.11 | |
| 1.3 | 1.1 | 1.7 | 1.8 | 1.8 | 1.6 |
| Rb2 | 0.09 | | 0.11 | | 0.12 | | 0.13 | | 0.13 | | 0.13 | |
| 0.7 | 2.5 | 1.8 | 2.0 | 1.8 | 1.9 |
| Rc | 0.11 | | 0.15 | | 0.15 | | 0.16 | | 0.17 | | 0.17 | |
| 2.7 | 4.7 | 4.7 | 4.1 | 4.1 | 4.3 |
| Re | 0.15 | | 0.23 | | 0.23 | | 0.24 | | 0.24 | | 0.24 | |
| 2.2 | 2.7 | 2.5 | 2.5 | 2.5 | 2.3 |
| Rd | 0.19 | | 0.27 | | 0.27 | | 0.28 | | 0.28 | | 0.28 | |
| 3.5 | 5.7 | 5.7 | 5.7 | 5.8 | 6.6 |
| Rg1 | 0.24 | | 0.37 | | 0.37 | | 0.37 | | 0.37 | | 0.38 | |
| 1.5 | 1.3 | 1.3 | 2.3 | 1.4 | 1.1 |
| Rf | 0.27 | | 0.39 | | 0.39 | | 0.41 | | 0.40 | | 0.40 | |
| | | | | |
Z ryciny 3 wynika, że analiza TLC ginsenozydów prowadzona w temperaturze poniżej 20°C oraz powyżej 28°C nie daje dobrych wyników rozdziału tych związków. Z wartości współczynników Rs przedstawionych w tabelach 6 i 7 wynika, że najlepszą rozdzielczość ginsenozydów w obu badanych układach rozwijających uzyskano dla temperatury 26°C. Wszystkie współczynniki Rs przekraczają wartość 1,5 co świadczy o bardzo dobrym rozdzieleniu badanych ginsenozydów. Z danych przedstawionych na rycinie 3 i w tabelach 6 oraz 7 wynika również, że temperatura rozwijania ma znacznie większy wpływ na rozdział ginsenozydów i ich wartości współczynników Rf w przypadku użycia układu (5) w porównaniu do układu (6). Chromatogramy TLC obrazujące rozdział wzorcowych ginsenozydów oraz ginsenozydów występujących w liściach i korzeniach żeń-szenia amerykańskiego przy użyciu fazy ruchomej (5) chloroform – octan etylu – metanol – woda, 15+40+22+9 (v/v) w 17 i 26°C przedstawia rycina 4.
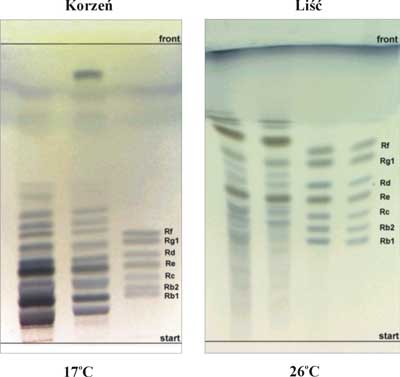
Ryc. 4. Chromatogramy TLC otrzymane w temperaturze 17 i 26°C przy użyciu fazy ruchomej nr 5.
Ocena jakościowa występowania ginsenozydów w częściach nadziemnych i podziemnych żeń-szenia pięciolistnego ( Panax quinquefolium L.) oraz preparatach farmaceutycznych
Badania TLC nad obecnością ginsenozydów w częściach nadziemnych (liście, łodygi i owoce) oraz podziemnych (korzenie) żeń-szenia amerykańskiego ( Panax quinquefolium L.) w I, II, III i IV roku wegetacji prowadzono na płytkach pokrytych żelem krzemionkowym Si 60G przy użyciu fazy ruchomej nr (5). Rozwijanie chromatogramów prowadzono w temp. 26°C. Chromatogramy oglądano w świetle widzialnym po wcześniejszej derywatyzacji odczynnikiem Godina.
Badania TLC wykazały, że w tych samych organach żeń-szenia, w różnym okresie rozwoju rośliny, występują te same ginsenozydy. Wyniki analizy jakościowej ginsenozydów występujących w korzeniach, liściach, łodygach i owocach roślin 4-letnich przedstawia tabela 8.
Tabela 8. Wyniki analizy TLC ginsenozydów występujących w częściach nadziemnych i podziemnych żeń-szenia pięciolistnego w IV roku wegetacji.
| Zidentyfikowany związek | Korzenie | Liście | Łodygi | Owoce |
Rb1
Rb2
Rc
Re
Rd
Rg1
Rf
Rg2 | ++
+/-
+
+
++
+
-
+ | +
+
+
+
++
+
-
++ | +
-
+
+
+
+
-
++ | -
+
-
+
+
+
-
++ |
| Intensywność barwy plamy: ++ duża; + średnia; +/- bardzo mała intensywność lub brak plamy; - związku nie wykryto |
W wyniku analizy TLC w korzeniach Panax quinquefolium zidentyfikowano 6 ginsenozydów: Rb1, Re, Rc, Rd, Rg1 i Rg2, w liściach 7 związków: Rb1, Rb2, Rc, Re, Rd, Rg1 i Rg2; w przypadku łodyg zidentyfikowano 6 związków: Rb1, Rc, Re, Rd, Rg1 i Rg2, natomiast w owocach 5 związków: Rb2, Re, Rd, Rg1 i Rg2. Z danych tych wynika, że związkami, które są obecne we wszystkich badanych organach żeń-szenia pięciolistnego, są ginsenozydy Re, Rd, Rg1 i Rg2. Analiza TLC wykazała również brak ginsenozydu Rf we wszystkich badanych organach Panax quinquefolium. Brak tego związku na chromatogramie TLC wg Farmakopei Europejskiej (59) odróżnia żeń-szeń pięciolistny od żeń-szenia właściwego.
W dalszej części pracy wykonano porównawczą analizę TLC ginsenozydów obecnych w P. quinquefolium i ginsenozydów występujących w dwu preparatach: Panaxan(r) i Extractum ginseng spir. spissum produkowanych z korzenia P. ginseng. Wyniki analizy przedstawia rycina 5.
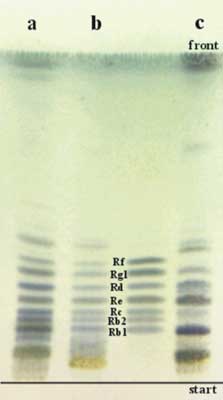
Ryc. 5. Chromatogram TLC ginsenozydów obecnych w Panax ginseng i Panax quinquefolium. a – Extractum ginseng spir. spissum; b – Panaxan(r); c – Panax quinquefolium (korzeń). Układ: Si 60G / chloroform – octan etylu – metanol – woda, 15+40+22+9 (v/v). Derywatyzacja: odczynnik Godina.
Z przeprowadzonych badań wynika, że metoda TLC jest szybką metodą pozwalającą na odróżnienie obu gatunków żeń-szenia. P. ginseng i P. quinquefolium różnią się nie tylko składem frakcji ginsenozydów, ale także zawartością poszczególnych związków, o czym świadczy intensywność poszczególnych pasm obecnych na chromatogramach obu gatunków żeń-szenia.
Wyniki analizy jakościowej cukrów i aglikonów powstałych po hydrolizie ginsenozydów
Analiza jakościowa ginsenozydów obejmowała rów-
nież badania TLC na obecność cukrów i aglikonów uwolnionych z połączeń glikozydowych w wyniku hydrolizy kwaśnej ginsenozydów. Badania na obecność cukrów prowadzono na płytkach pokrytych celulozą w układzie: n-butanol – pirydyna – woda, 6+4+3 (v/v) rozwijając płytkę dwukrotnie na tym samym dystansie (54, 55). Tak rozwinięte płytki derywatyzowano ftalanem aniliny. Wyniki analizy TLC cukrów uwolnionych w wyniku hydrolizy kwaśnej frakcji ginsenozydów wyodrębnionych z liści i korzeni żeń-szenia pięciolistnego przedstawia tabela 9.
Tabela 9. Wyniki analizy TLC cukrów uwolnionych w wyniku hydrolizy ginsenozydów.
| Zidentyfikowany cukier | Wartości współczynników Rf | Barwa plamy (po derywatyzacji ftalanem aniliny) |
| Glukoza | 0,571 | brązawo-szara |
| Arabinoza | 0,629 | ceglasto-różowa |
| Ksyloza | 0,714 | ceglasto-różowa |
| Ramnoza | 0,836 | brązawo-szara |
Z danych zawartych w tabeli 9 wynika, że w hydrolizacie zidentyfikowano 4 związki. Były to: glukoza, arabinoza, ksyloza i ramnoza. Porównanie intensywności zabarwienia poszczególnych plam obecnych na chromatogramach TLC wskazuje na dużą, w porównaniu do pozostałych cukrów, zawartość glukozy. Z tego wynika, że glukoza jest głównym cukrem wchodzącym w skład cząsteczek ginsenozydów. Obecność na chromatogramach TLC plam arabinozy, ksylozy i ramnozy potwierdza występowanie w badanych surowcach (liściach i korzeniach) ginsenozydów: Rc, Re, Rb2 i Rg2, a także może wskazywać na obecność innych ginsenozydów (Ra1, Ra2, Ras, Rbs, Rs1, Rs2), których identyfikacja nie była możliwa ze względu na brak wzorców.
Badania na obecność aglikonów prowadzono na płytkach żelowych w dwóch układach: chloroform – metanol, 10+1 (v/v) (52) i benzen – octan etylu, 1+1 (v/v) (53). Otrzymane chromatogramy wywoływano przez spryskanie odczynnikiem Godina. Analiza jakościowa aglikonów uwolnionych w wyniku hydrolizy ginsenozydów wskazała na obecność kwasu oleanolowego. Występowanie tego związku wskazuje na obecność w obu badanych surowcach ginsenozydów pochodnych kwasu oleanolowego, np. ginsenozydu Ro, czy chikusetsaponiny IVa. Oprócz kwasu oleanolowego na chromatogramach TLC obecne były również inne plamy należące prawdopodobnie do protopanaxadiolu i protopanaxatriolu, a także panaxadiolu i paraxatriolu powstałych w wyniku cyklizacji dwóch poprzednich (69). Identyfikacja tych związków była niemożliwa ze względu na brak wzorców.
Wyniki analizy jakościowej ginsenozydów metodą OPLC
Cienkowarstwowa chromatografia ciśnieniowa (OPLC) jest techniką, którą charakteryzuje wymuszony przepływ fazy ruchomej, a proces rozdziału badanych związków prowadzony jest w całkowicie zamkniętych komorach niewysyconych (60-63). Takie warunki rozdziału chromatograficznego powodują wzrost współczynników retencji chromatografowanych substancji w porównaniu do współczynników retencji otrzymanych w wyniku rozdziału substancji przy zastosowaniu klasycznej chromatografii cienkowarstwowej (63-65). Zastosowanie do analizy OPLC fazy ruchomej stosowanej do rozwijania związków metodą TLC jest możliwe, gdy wartości współczynników Rf substancji nie przekraczają wartości 0,8 (64). Zmiana techniki rozwijania z liniowej na kołową powoduje, że wartości współczynników Rf rozwijanych substancji rosną następująco: Rflin = (Rfcirc)2 (66, 67).
Badania nad rozdziałem ginsenozydów obecnych w korzeniach i liściach żeń-szenia pięciolistnego metodą chromatografii OPLC rozpoczęto od doboru odpowiedniej fazy ruchomej. Punktem wyjścia były dwie fazy ruchome dające najlepsze wyniki rozdziału ginsenozydów przy zastosowaniu klasycznej chromatografii cienkowarstwowej, tj. (5) chloroform – octan etylu – metanol – woda, 15+40+22+9 ( v/v) oraz (6) chloroform – octan etylu – metanol – woda – heksan, 20+60+22+8+4 ( v/v). Ponadto modyfikowano fazę ruchomą nr (6) zmieniając octan etylu na octan metylu, propylu i butylu (faza ruchoma (7), (8) i (9).
Porównanie wyników rozdziału ginsenozydów metodami TLC, HPTLC, i OPLC przy użyciu fazy ruchomej nr (5) przedstawia rycina 6.
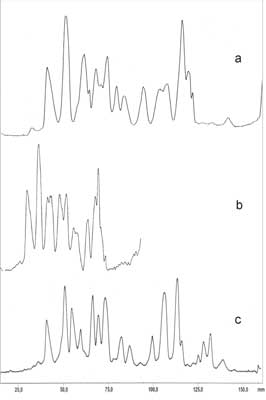
Ryc. 6. Rozdział ginsenozydów występujących w korzeniach P. quinquefolium przy użyciu fazy ruchomej (5): chloroform – octan etylu – metanol – woda, 15+40+22+9 (v/v). a – TLC; b – HPTLC; c – OPLC.
Z ryciny 6 wynika, że zastosowanie różnych metod rozwijania znacząco wpływa na rozdział frakcji ginsenozydów. Najbardziej czułą metodą jest metoda OPLC. Rozwijanie metodą OPLC prowadzono na płytkach HPTLC na dystansie 155 mm. Dalsze zwiększenie dystansu rozwijania nie wpływało na rozdzielczość pików; rosły jedynie wartości współczynników Rf poszczególnych plam.
Jak wykazały analizy TLC i HPTLC (ryc. 7), dobre wyniki rozdziału ginsenozydów uzyskano dla fazy ruchomej nr (6): chloroform – octan etylu – metanol – woda – heksan, 20+60+22+8+4 ( v/v).
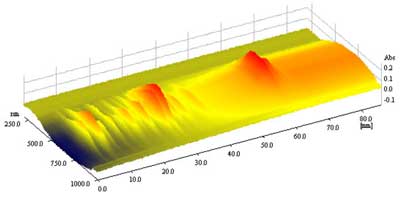
Ryc. 7. 3D-chromatogram otrzymany w wyniku rozwijania frakcji ginsenozydów z korzenia żeń-szenia w układzie chloroform – octan etylu – metanol – woda – heksan, 20+60+22+8+4 (v/v) na płytkach HPTLC.
Badano również, jaki wpływ na rozdział ginsenozydów ma zmiana octanu etylu, wchodzącego w skład fazy nr (6), na jeden z jego homologów: octan metylu (Me-ac), propylu (Pr-ac) i butylu (Bu-ac).
Zależność pomiędzy wartościami współczynników Rm dla poszczególnych ginsenozydów, a modyfikatorem fazy ruchomej dla analizy TLC i HPTLC, przedstawia rycina 8.
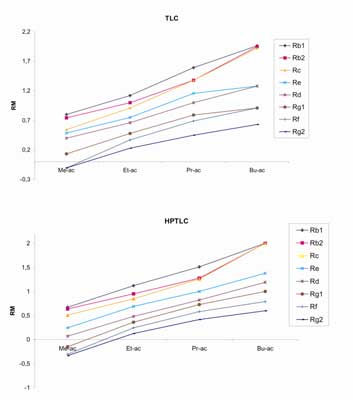
Ryc. 8. Zależność wartości współczynników Rm dla wzorcowych ginsenozydów od rodzaju modyfikatora fazy ruchomej dla metod: TLC – A oraz HPTLC – B.
Jak wynika z ryciny 8, najlepszy rozdział ginsenozydów, zarówno na płytkach TLC, jak i HPTLC, uzyskano przy zastosowaniu fazy ruchomej zawierającej w swoim składzie octan etylu. Dobrą selektywność otrzymano również dla układu zawierającego w swoim składzie octan propylu, jednakże rozdział dwóch ginsenozydów (Rb2 i Rc) był niemożliwy zarówno dla metody TLC jak i HPTLC.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Wolski T, Ludwiczuk A, Baj T i wsp. Rodzaj Panax - systematyka, skład chemiczny, działanie i zastosowanie oraz analiza fitochemiczna nadziemnych i podziemnych organów żen-szenia amerykańskiego - Panax quinquefolium L.). Cz. I. Postępy Fitoterapii 2008; 2:96-114. 2. Ando T, Tanaka O, Shibata S. Chemical studies on the oriental plant drugs. XXV. Comparative studies on the saponins and sapogenins of ginseng and related crude drugs. Svoyakugaku Zasshi 1971; 25:28-32. 3. Chuang WC, Sheu SJ. Determination of ginsenosides in ginseng crude extracts by high-performance liquid chromatography. J Chromatogr A 1994, 685:243-51. 4. Wu J, Lin L, Chau F. Ultrasound-assisted extraction of ginseng saponins from ginseng roots and cultured ginseng cells. Ultrason Sonochem 2001; 8:347-52. 5. Choi JH, Kim DH, Sung HS i wsp. Kinetic studies on the thermal degradation of ginsenosides in ginseng extract. Korean J Food Sci Tech1982; 14(3):197-202. 6. Sung HS, Yang JW. Effect of the heating treatment on the stability of saponin in white ginseng. J. Korean Society of Food and Nutrition 1986; 15:22-6. 7. Mason TJ, Paniwnyk L, Lorimer JP. The uses of ultrasound in food technology. Ultrason Sonochem 1996; 3:253-60. 8. Giergielewicz-Możajska H, Dąbrowski Ł, Namieśnik J. Przegląd technik ekstrakcyjnych wykorzystywanych na etapie wyodrębniania analitów z próbek stałych. Ekologia i Technika 2001; 1(49):3-11. 9. Choi MPK, Chan KKC, Leung HW i wsp. Pressurized liquid extraction of active irgendients (ginsenosides) from medicinal plants using non-ionic surfactant solutions. J Chromatogr A, 2003; 983:153-62. 10. Lopez-Avila V, Young R, Teplitsky N. (1996), Microvawe-assisted extraction as an alternative to Soxhlet, sonication and supercritical fliud extraction. Journal of the Association of Official Analytical Chemists International 1996; 79(1):142-56. 11. Kwon JH, Belanger JMR, Pare JRJ i wsp. Application of the microwave-assisted process (MAPTM) to the fast extraction of ginseng saponins. Food Research International 2003; 36:491-8. 12. Wolski T, Ludwiczuk A. (2001), Ekstrakcja produktów naturalnych gazami w stanie nadkrytycznym. Przem Chem 2001; 80 (7):286-9. 13. Wang J, Sha Y, Li W i wsp. Quinquenoside L9 from leaves and stems of Panax quinquefolium L. J Asian Nat Prod Res 2001; 3(4):293-7. 14. Sticher O, Soldati F. HPLC Trennung und quantitative Bestimmung der Ginsenoside von Panax ginseng, Panax quinquefolium und von Ginseng-Spezialitäten. Planta Med 1979; 36:30-42. 15. Glockl I, Veit M, Blaschke G. Determination of ginsenosides from Panax ginseng using micellar electrokinetic chromatography. Planta Med 2002; 68(2):158-61. 16. Hiai S, Oura H, Hamanaka H i wsp. A color reaction of panaxadiol with vanillin and sulphuric acid. Planta Med 1975; 28:131-8.17. Hiai S, Oura H, Odaka Y i wsp. A colorimetric estimation of ginseng saponins. Planta Med 1975a; 28:363-9. 18. Farmakopea Niemiecka DAB 10, Ginseng radix, 1991. 19. Hostettmann K, Terreaux C, Marston A i wsp. The role of Planar Chromatography in the rapid screening and isolation of bioactive compounds from medicinal plants. J Planar Chromatogr 1997; 10:251-7. 20. Corthout J, Naessens T, Apers S i wsp. Quantitative determination of ginsenosides from Panax ginseng roots and ginseng preparations by thin layer chromatography-densitometry. J Pharm Biomed Anal 1999; 21:187-92. 21. Vanhaelen-Fastre RJ, Faes ML, Vanhaelen MH. High-performance thin-layer chromatographic determination of six major ginsenosides in Panax ginseng. J Chromatogr A 2000; 868:269-76. 22. Nyiredy Sz. Rotation Planar Chromatography. In: Sz. Nyiredy (Editor) Planar Chromatography. A retrospective view for the third millennium. Springer, Budapest, 2001; 178-99. 23. Ludwiczuk A, Nyiredy Sz, Wolski T. Separation of the ginsenosides fraction obtained from the roots of Panax quinquefolium L. cultivated in Poland. J Planar Chromatogr 2005; 2:104-7. 24. Ludwiczuk A, Kołodziej B, Wolski T. Zawartość i skład ginsenozydów w różnych organach żeń-szenia pięciolistnego ( Panax quinquefolium L.) Acta Agrobot 2006; 59(1):507-14. 25. Ligor T, Ludwiczuk A, Wolski T, Buszewski B. Isolation and determination of ginsenosides in American ginseng leaves and root extracts by LC-MS. Anal Bioanal Chem 2005; 383(7-8):1098-105. 26. Wang X, Sakuma T, Asafu-Adjaye E i wsp. Determination of ginsenosides in plant extracts from Panax ginseng and Panax quinquefolium by LC/MS/MS. Anal Chem 1999; 71(8):1579-84. 27. Shangguan D, Han H, Zhao R i wsp. New method for high-performance liquid chromatographic separation and fluorescence detection of ginsenosides. J Chromatogr A 2001; 910:367-72. 28. Cui JF. Identification and quantification of ginsenosides in various commercial ginseng preparations. European J Pharm Sci 1995; 3:77-85. 29. Cui JF, Bjorkhem I, Eneroth P. Gas chromatographic - mass spectrometric determination of 20(S)-protopanaxadiol and 20(S)-protopanaxatriol for study on human urinary excretion of ginsenosides after ingestion of ginseng preparations. J Chromatogr B 1997; 689:349-55. 30. Kubo M, Tani T, Katsuki T i wsp. (1980), Histochemistry. I. Ginsenosides in ginseng ( Panax ginseng C.A. Meyer). J Nat Prod 1980; 43(2):278-84. 31. Segiet-Kujawa E, Lutomski J. Comparison of analytical methods of determining saponins in some Araliaceae species. Herba Pol 1986; 32(1):39-46. 32. Sticher O. Getting to the root of ginseng. CHEMTECH 1998; 28(4):26-32. 33. Dou DQ, Hou WB, Chen YJ. Studies on the characteristic constituents of Chinese ginseng and American ginseng. Planta Med 1998; 64:585-6. 34. Ludwiczuk A, Wolski T, Berbeć S. Chromatographic analysis of ginsenosides occurring in roots of American ginseng ( Panax quinquefolium L.) and Asian ginseng ( Panax ginseng C.A. Mayer) preparations. J Planar Chromatogr 2002; 15:147-50. 35. Dallenbach-Toelke K, Nyiredy Sz, Meszaros SY i wsp., Sticher O. TLC, HPTLC and OPLC separation of ginsenosides. J High Res Chromatogr & Chromatogr Comm 1987; 10:362-4. 36. Soldati F, Sticher O. HPLC separation and quantitative determination of ginsenosides from Panax ginseng, Panax quinquefolium and from Ginseng Drug Preparations. Planta Med 1980; 38:346-57. 37. Court WA, Hendel JG, Elmi J. Reversed-phase high-performance liquid chromatographic determination of ginsenosides of Panax quinquefolium. J Chromatogr A 1996; 775:11-7. 38. Kwon SW, Han SB, Park IH i wsp. Liquid chromatographic determination of less polar ginsenosides in processed ginseng. J Chromatogr A 2001; 921:335-9. 39. Park MK, Kim BK, Park JH i wsp. High-performance liquid-chromatographic determination of ginsenosides using photoreduction fluorescence detection. J Liq Chromatogr, 1995; 18(10):2077-88. 40. Samukawa K, Yamashita H, Matsuda H i wsp. Simultaneous analysis of ginsenosides of various Ginseng radix by HPLC. Yakugaku-Zasshi 1995; 115(3):241-9. 41. Zhou Z, Zhang G. Analysis of ginseng. IV. HPLC determination of ginsenosides in Panax ginseng. Youxue-Xuebao 1988; 23(2):137-41. 42. Park MK, Park JH, Han SB i wsp. High-performance liquid-chromatographic analysis of ginseng saponins using evaporative light-scattering detection. J Chromatogr A 1996; 736(1-2): 77-81. 43. Ji HY, Lee HW, Kim HK i wsp. Simultaneous determination of ginsenoside Rb1 and Rg1 in human plasma by liquid chromatography - mass spectrometry. J Pharm Biomed Anal 2004; 35:207-12. 44. Lau AJ, Woo SO, Koh HL. Analysis of saponins in raw and steamed Panax notoginseng using high-performance chromatography with diode array detection. J Chromatogr A 2003; 1011:77-87. 45. Tani T, Kubo M, Katsuki T i wsp. Histochemistry. II. Ginsenosides in ginseng ( Panax ginseng, root). J Nat Prod 1981; 44(4):401-7. 46. Farmakopea Polska V, tom 1, Supl. PTFarm, Warszawa, 1995. 47. Farmakopea Polska VI, PTFarm, Warszawa, 2002. 48. Metoda Badawcza Nr MB:05F1100202, ZZ Herbapol w Klęce S.A.: Oznaczanie zawartości ginsenozydów w preparacie Extractum Ginseng spir. spiss. 49. Ludwiczuk A. Badania składu chemicznego w ontogenezie żeń-szenia amerykańskiego Panax quinquefolium L.. Praca doktorska. Uniwersytet Medyczny, Lublin, 2005. 50. Ludwiczuk A, Wolski T. Estimation of the content and composition of ginsenosides occurring in extracts from American ginseng and Asian ginseng. Annales UMCS, sec. EEE 2003; 7:53-8. 51. Ouyang MA. Glycosides from the leaves of Ilex latifolia. Chinese J Chem 2001; 19:885-92. 52. Strzelecka H, Kamińska J, Kowalski J i wsp. Chemiczne metody badań roślinnych surowców leczniczych. PZWL, Warszawa, 1987; pp. 312. 53. Nguyen TN. Study on Panax vietnamensis Ha et Grushv. - Araliaceae. Botany - tissue culture. Chemistry - biological properties. Herba Pol 1989; 35, Supl.II: pp.229. 54. Jerzmanowska Z. Substancje roślinne. Metody wyodrębniania. Tom II. PWN, Warszawa, 1970; pp. 200. 55. Gudej J, Tomczyk M, Urban E i wsp. Badania składu chemicznego liści Rubus saxatilis L. Herba Pol 1998; 44 (4):340-4. 56. Sieńko A. Dobór warunków ekstrakcji i rozdziału chromatograficznego ginsenozydów żeń-szenia amerykańskiego Panax quinquefolium L. Praca magisterska, Uniwersytet Medyczny w Lublinie, 2003; 84pp. 57. Ziewiec A. Badania nad składem chemicznym części nadziemnych i podziemnych żeń-szenia amerykańskiego Panax quinquefolium L. Praca magisterska, Uniwersytet Medyczny w Lublinie, 2002; 87pp . 58. Ludwiczuk A, Wolski T, Nyiredy Sz. Circular and linear OPLC of ginsenosides in Panax quinquefolium L. cultivated in Poland. Proceedings of the International Symposium on Planar Separations, Budapeszt, 21-23 czerwiec 2003, Węgry, 291-6. 59. Farmakopea Europejska, European Pharmacopoeia - Supl. 2001; 887-9. 60. Tyihak E, Mincsovics E. Forced-flow Planar Liquid Chromatographic Techniques. J Planar Chromatogr 1988; 1:6-19. 61. Tyihak E, Mincsovics E. Trends in Overpressured Layer Chromatography. J Planar Chromatogr 1991; 4:288-92. 62. Tyihak E, Mincsovics E. Overpressured-layer chromatography (optimum performance laminar chromatography). In: Sz. Nyiredy (Editor) Planar Chromatography. A retrospective view for the third millennium. Springer, Budapest, 2001; 137-76. 63. Nyiredy Sz. The bridge between TLC and HPLC: overpressured layer chromatography (OPLC). Trends in Analytical Chemistry 2001; 20 (2):91-101. 64. Nyiredy Sz. Planar chromatographic method development using the PRISMA optimization system and flow charts. J Chromatogr Sci 2002; 40:1-11. 65. Nyiredy Sz, Fater Zs, Botz L i wsp. The role of chamber saturation in optimization of planar chromatography. J Planar Chromatogr 1992; 5:308-15. 66. Dallenbach-Toelke K, Sticher O. Comparison of Circular and Linear Overpressured Layer Chromatography (OPLC). J Planar Chromatogr 1988; 1:73-5. 67. Szabady B. The different modes of development. In: Sz. Nyiredy (Editor). Planar Chromatography. A retrospective view for the third millennium. Springer, Budapest, 2001; 88-102. 68. Nyiredy Sz, Meszaros SY, Dallenbach-Toelke K i wsp. The "Disturbing Zone" in Overpressure Layer Chromatography (OPLC). J High Res Chromatogr and Chromatogr Comm 1987; 10:352-6. 69. Shibata S, Tanaka O, Shojii J i wsp. Chemistry and pharmacology of Panax. In H. Wagner, H. Hikkino and N.R. Farnsworth (Eds.) Economic and Medicinal Plant Research, Vol.1. Orlando, Fla: Academic Press, 1985; 218-84.
otrzymano: 2008-08-21
zaakceptowano do druku: 2008-09-05
Adres do korespondencji:
*Tadeusz Wolski
Katedra i Zakład Farmakognozji z Pracownią Roślin Leczniczych Uniwersytetu Medycznego w Lublinie
ul. Chodźki 1, 20-093 Lublin
tel.: (0-81) 742-38-10, fax: (0-81) 742-38-09
e-mail: twolski@pharmacognosy.org
Postępy Fitoterapii 3/2008Strona internetowa
czasopisma Postępy FitoterapiiPozostałe artykuły z numeru 3/2008:
