© Borgis - Postępy Nauk Medycznych 2/2008, s. 132-137
*Lucyna Papierska
Pierwotna autoimmunologiczna niedoczynność kory nadnerczy
Autoimmune primary adrenal insufficiency
Klinika Endokrynologii Centrum Medycznego Kształcenia Podyplomowego w Warszawie
Kierownik Kliniki: prof. CMKP dr hab. med. Wojciech Zgliczyński
Streszczenie
Autoimmunologiczna pierwotna niedoczynność kory nadnerczy (Choroba Addisona = pnkn) to endokrynna choroba autoimmunologiczna, której istotą jest spowodowana procesem autoimmunizacyjnym destrukcja kory nadnerczy. Jest chorobą rzadką – występuje z częstością około 100 przypadków na milion populacji, ale poważną – nierozpoznana i nieleczona prowadzi do śmierci. Często współwystępuje z innymi chorobami układu wewnątrzwydzielniczego o podłożu autoimmunologicznym, na przykład autoimmunologicznymi chorobami tarczycy, cukrzycą typu I i przedwczesnym wygasaniem czynności jajników. W stadium przedklinicznym w surowicy chorych wykrywane są przeciwciała przeciw 21-hydroksylazie i innym enzymom szlaku syntezy steroidów. Mogą one służyć za wczesny wskaźnik ryzyka rozwoju pnkn. Objawy kliniczne choroby Addisona są najczęściej niespecyficzne – mogą być nimi na przykład męczliwość, nudności, bóle brzucha i chudnięcie. Dlatego też choroba często rozpoznawana jest dopiero wtedy, gdy rozwinie się zagrażający życiu przełom nadnerczowy. W leczeniu pnkn stosowane są hydrokortyzon, fludrokortyzon i dehydroepiandrosteron. Prawidłowo prowadzona terapia substytucyjna zmniejsza objawy choroby, ale przedawkowanie leków może prowadzić do rozwoju osteoporozy, insulinooporności i chorób układu krążenia.
Summary
Autoimmune primary adrenal insufficiency (Addison´s disease – AAD) is an endocrine and immunological disease, caused by the autoimmune destruction of the adrenal cortex. The disease is rare – about 100 cases per million inhabitants, but serious – if misdiagnosed and untreated it leads to death. AAD frequently concurs with other autoimmune endocrine diseases, such as autoimmune thyroid diseases, type 1 diabetes mellitus, and premature ovarian failure. In the preclinical stage of ADD, antibodies against 21-hydroxylase and other enzymes in the pathway of steroid synthesis are detected. They may serve as the earliest indicator of the risk of the disease. The most frequent symptoms and signs of overt Addison´s disease such as fatigue, nausea, abdominal pain and weight loss are largely unspecific. Therefore diagnosis is frequently established only after a life-threatening adrenal crisis has occurred. Hydrocortisone, fludrocortisone and dehydroepiandrosterone are hormones used in the treatment of the Addison´s disease. Correct replacement therapy usually alleviates the symptoms of ADD, but if overdosed may lead to osteoporosis, insulin resistance and cardiovascular diseases.

Wstęp
Pierwotną niedoczynność kory nadnerczy czyli chorobę Addisona (pnkn) zidentyfikowano i opisano już w połowie XIX wieku (1). Chorobę mogą powodować infekcje, choroby spichrzeniowe, przerzuty nowotworowe do nadnerczy, rzadkie choroby o podłożu genetycznym (tab. 1). Przez ponad stulecie jej najczęstszą przyczyną była gruźlica nadnerczy. W ostatnim dwudziestoleciu dominuje jednak niedoczynność o podłożu autoimmunologicznym, która występuje w różnych populacjach z częstością 93-140 na milion, a nowe przypadki stanowią około 5/milion na rok (2). Najwyższą częstość jej występowania opisano w Norwegii – 140/milion (3). Opierając się na powyższych danych można przypuszczać, że w Polsce na pierwotną niedoczynność nadnerczy choruje co najmniej 4000 osób, a co roku możemy spodziewać się około 200 nowych rozpoznań tej choroby.
Tabela 1. Przyczyny pierwotnej niedoczynności kory nadnerczy.
| | Częstość występowania |
a. O podłożu autoimmunologicznym
1. izolowana,
2. W przebiegu zespołów wielogrucołowych
(APS I, APS II)
b. W przebiegu infekcji (Tbc, grzybica, CMV, AIDS)
c. Obustronne przerzuty nowotworowe
d. Nacieki w chorobach spichrzeniowych (amyloidoza, hemochromatoza)
e. Obustronne wylewy do nadnerczy
f. Adrenoleukodystrofia
g. Wrodzona hipoplazja nadnerczy
h. Niewrażliwość na ACTH (zespół trzech A, rodzinny niedobór glikokortykoidów)
i. stan po obustronnej adrenalektomii | 70-90% |
Nieleczona pnkn jest chorobą śmiertelną. Do momentu zsyntetyzowania kortyzonu, a następnie jego pochodnych 2 lata od rozpoznania przeżywał co piąty chory. W latach 60-tych XX wieku wydawało się, że przeżywalność pacjentów leczonych glukokortykoidami oraz jakość ich życia nie są gorsze niż zdrowej populacji (4). Europejskie raporty z ostatnich kilkunastu lat podważają jednak te optymistyczne stwierdzenia, wykazując zwiększoną śmiertelność wśród chorych z chorobą Addisona i gorszą jakość ich życia (5, 6). Wydaje się, że za powikłania choroby może odpowiadać zarówno niedostateczne wyrównanie niedoboru hormonów nadnerczowych, jak i nieuzasadnione zwiększanie ich dawek. Nie opracowano do dziś obiektywnych metod oceny stopnia wyrównania niedoborów hormonalnych podczas leczenia substytucyjnego (7).
Rozpoznanie pierwotnej niedoczynności kory nadnerczy – jak wcześnie jesteśmy w stanie wykryć zagrożenie?
Nierozpoznana i nieleczona pierwotna niedoczynność kory nadnerczy jest chorobą śmiertelną, a wydaje się, że zdiagnozowane już przypadki choroby to jedynie „wierzchołek góry lodowej”. Choroba Addisona u połowy chorych rozpoznawana jest dopiero w momencie wystąpienia jej najgroźniejszego powikłania, czyli przełomu nadnerczowego (8). U połowy pacjentów po wyleczeniu przełomu stwierdzano, że pierwsze objawy pojawiły się już co najmniej rok przed hospitalizacją. Są one mało specyficzne, sugerujące chorobę przewodu pokarmowego (bóle brzucha, brak apetytu, nudności i wymioty, utrata masy ciała), pierwotną chorobę mięśni (bóle mięśni, miastenia), depresję (osłabienie, obniżony nastrój i napęd). Najczęściej ciśnienie tętnicze krwi obniża się, ale może występować jedynie hipotonia ortostatyczna. Warto pamiętać, że stwierdzenie nadciśnienia tętniczego nie wyklucza niedoczynności kory nadnerczy, choć wówczas hipotonię ortostatyczną należy różnicować z efektem działania leków hipotensyjnych. Ciemnienie skóry, charakterystyczne dla niedoczynności pierwotnej może w pierwszych miesiącach choroby być bardzo dyskretne, albo maskowane przez współwystępujące z pnkn bielactwo. W badaniach dodatkowych krwi często stwierdzamy hiponatremię i hiperkaliemię, umiarkowanie podwyższone stężenie TSH, czasem hiperkaliemię (9, 10, 11,12). Zestawienie najczęściej stwierdzanych objawów przedmiotowych i podmiotowych oraz odchyleń w badaniach dodatkowych przedstawiono w tabeli 2.
Tabela 2. Objawy przedmiotowe, podmiotowe i wyniki badań dodatkowych w pierwotnej niedoczynności kory nadnerczy.
| | Częstość występowania |
Objawy podmiotowe:
a. osłabienie, zmęczenie,
b. brak apetytu
c. objawy ze strony przewodu pokarmowego
- nudności
- wymioty
- obstrukcje
- bóle brzucha
- biegunka
d. łaknienie soli
e. zawroty głowy
f. bóle mięśni i stawów | 100%
100%
92%
86%
75%
33%
31%
6%
16%
12%
6-13% |
Objawy przedmiotowe:
a. utrata ciężaru ciała
b. ciemne zabarwienie skóry
c. niedociśnienie (RR skurczowe < 110 mm Hg)
d. bielactwo | 100%
94%
88-94%
10-20% |
Zmiany w wynikach rutynowych badań krwi
a. zaburzenia elektrolitowe
- hiponatremia
- hiperkaliemia
- hiperkalcemia
b. azotemia
c. anemia
d. eozynofilia | 92%
88%
64%
6%
55%
40%
17% |
Przy uzasadnionym podejrzeniu niedoczynności kory nadnerczy warto włączyć leczenie Hydrokortyzonem nawet już przed wysłaniem chorego do specjalisty. Pozwala to uniknąć zagrożenia przełomem, a nie przeszkadza w sposób znaczący w dalszej diagnostyce. Jeżeli rozpoznanie jest prawidłowe, poprawę stanu pacjenta można zauważyć już w pierwszych dniach stosowania Hydrokortyzonu. Wystarczy doba przerwy w stosowaniu leku, aby wykonać miarodajne oznaczenia stężenia kortyzolu i ACTH w surowicy, a dwie doby aby przeprowadzić dobową zbiórkę moczu z oznaczeniem dobowego wydalania kortyzolu lub 17hydroksykortykoidów (17OHCS). W wypadku obawy, że stan pacjenta może ulec pogorszeniu na skutek przerwy w leczeniu Hydrokortyzonem, należy zastosować osłonę z Deksametazonu (2 x 0,25 mg), z opcjonalnie dołączonym fludrokortyzonem. W przypadku niedoczynności pierwotnej, w której uszkodzona jest kora, czyli efektor dla działania ACTH rozpoznanie można łatwo potwierdzić wykonując test z syntetycznym 1-24ACTH (Synacthen, Cortrosyn) (13). (Proponowany schemat postępowania diagnostycznego – patrz ryc. 1).
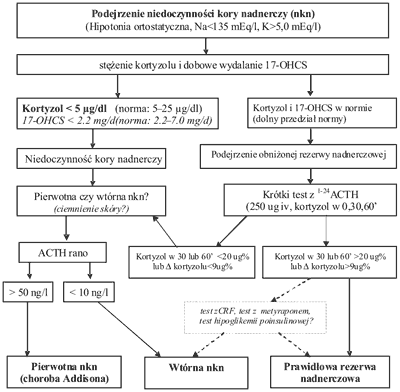
Ryc. 1. Propozycja postępowania diagnostycznego w przypadku podejrzenia niedoczynności kory nadnerczy.
Prawdopodobnie dość liczną grupą chorych niezdiagnozowanych są osoby z tak zwaną subkliniczną pierwotną niedoczynnością kory nadnerczy. Stwierdzono u nich podwyższone miano przeciwciał przeciw 21-hydroksylazie, enzymowi kluczowemu dla zachodzącej w warstwie pasmowatej syntezy kortyzolu. W tej grupie może występować podwyższone stężenie ACTH, opisywano też zwiększoną aktywność reninową osocza. Test stymulacji 250 μg 1-24 ACTH nie wykazuje nieprawidłowości, ale według niektórych badaczy można stwierdzić niedostateczny przyrost kortyzolu po 1 μg Synacthenu (14). Inne przeciwciała obecne w surowicy w przypadkach subklinicznej niedoczynności kory nadnerczy to przeciwciała przeciw desmolazie cholesterolowej SCC i 17α-hydroksylazie (17OH), choć częściej są one stwierdzane w rzadko występującym autoimmunologicznym zespole wielogruczołowym typu I (APS-1) (15).
W wypadku subklinicznej pnkn nie stwierdza się jakichkolwiek objawów choroby. Jednak co najmniej połowa osób z podwyższonym mianem przeciwciał przeciwnadnerczowych w ciągu dziesięciu lat od ich wykrycia rozwija objawową niedoczynność kory nadnerczy (16). Rozpoznanie stadium przedklinicznego choroby może mieć ogromne znaczenie dla zapobiegania wystąpieniu przełomu nadnerczowego, którego ryzyko wzrasta w miarę rozwoju choroby. Identyfikacja genów odpowiedzialnych za rozwój procesu autoimmunizacyjnego z niszczeniem kory nadnerczy pozwoli w przyszłości wyodrębnić osoby narażone na rozwój choroby (opisano kilka genów-kandydatów) (17,18).
W ponad 50% przypadków, niedoczynności kory nadnerczy towarzyszą choroby autoimmunologiczne innych narządów endokrynnych (autoimmunologiczne zapalenie tarczycy, przedwczesne wygasanie czynności jajników, cukrzyca typu I.) oraz inne choroby z autoimmunizacji (bielactwo, anemia z niedoboru B12, celiakia). Powyższe zaburzenia łączą się w autoimmunologiczne zespoły wielogruczołowe – aps ( autoimmune polyglandular syndromes). Ponieważ oznaczenie miana przeciwciał przeciw 21-hydroksylazie w całej populacji nie jest możliwe, a badania genetyczne to na razie przyszłość, badaniem tym powinno się objąć przynajmniej chorych na wymienione powyżej choroby oraz ich rodziny.
Leczenie pierwotnej niedoczynności kory nadnerczy
Istotą leczenia pierwotnej niedoczynności kory nadnerczy jest uzupełnienie niedoborów glukokortykoidów, mineralokortykoidów i, opcjonalnie, androgenów nadnerczowych.
Glukokortykoidy
Lekiem z wyboru w leczeniu niedoczynności kory nadnerczy, jest Hydrokortyzon (czyli syntetyczny kortyzol). W niektórych krajach stosowany jest octan kortyzonu, którego aktywność glukokortykoidowa zależy od aktywności 11β-dehydrogenazy steroidowej typu 1. Leczenie prednizolonem lub deksametazonem jest nieuzasadnione z patofizjologicznego punktu widzenia – steroidy te mają długi okres półtrwania i działają w godzinach nocnych, co zwiększa ryzyko powikłań hiperkortyzolemii. Dobowa produkcja kortyzolu przez zdrowe nadnercza wynosi 5 do 10 mg/m2 powierzchni ciała. Odpowiada to dawce 15-25 mg Hydrokortyzonu, albo 25-37,5 mg octanu kortyzonu. Po zażyciu Hydrokortyzonu występuje szybki przyrost stężenia kortyzolu w surowicy, a następnie gwałtowne jego obniżanie się, tak że 5-8 godzin po zażyciu tabletki kortyzolemia nie przekracza 4 μg%. Hydrokortyzon powinien być stosowany w trzech dawkach: porannej, południowej i popołudniowej. W razie stosowania dwóch dawek (dopuszczalna, choć gorsza opcja), druga powinna być zażyta nie później niż 6-7 godzin po pierwszej. Leku nie stosuje się na noc! (9-13, 19).
Indywidualna wrażliwość na hydrokortyzon zależna jest od procesu wchłaniania leku, gęstości receptorów dla glukokortykoidów, aktywności 11βHSD oraz wrażliwości receptora (opisano dwa polimorfizmy, zwiększające wrażliwość na glukokortykoidy) (20, 21). Ponieważ w codziennej praktyce niemożliwa jest ocena tych wszystkich parametrów, dawkę dobieramy najczęściej w sposób empiryczny.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Addison T: On the constitutional and local effects of disease of the supra-renal capsules. London: Samuel Highley, 1855; viii, 4to 43.
2. Laureti S, et al.: Is the prevalence of Addison´s disease underestimated? J. Clin. Endocrinol. Metab., 1999; 84: 1762.
3. Lovas K, et al.: High prevalence and increasing incidence of Addison´s disease In western Norway. Clin. Endocrinol., (Oxf). 2002; 56:787-791.
4. AS Mason, et al.: Epidemiological and clinical picture of Addison´s disease Lancet. 1968; 5; 2(7571): 744-7.
5. Bergthorsdottir R, et al.: Premature Mortality in Patients with Addison´s Disease: A Population-Based Study. The Journal of Clinical Endocrinology & Metabolism 2006; 91(12): 4849-4853.
6. Lovas K, et al.: Subjective health status in Norwegian patients withAddison´s disease. Clin Endocrinol (Oxf). 2002; 56:581-588.
7. Lukert BP: Glucocorticoid Replacement – How Much Is Enough? The Journal of Clinical Endocrinology & Metabolism 2006; 91(12): 3793-794.
8. Zelissen PM: Addison patients in the Netherlands. Medical report of the survey. The Hague, NL: Dutch Addison Society, 1994.
9. Stewart PM: Glucocorticoid deficiency w: Wiliams Textbook of Endocrinology 2003; s. 525-532.
10. Arlt W, Allolio B: Adrenal insufficiency. Lancet 2003; 361: 1881-93.
11. Papierska L. Niedoczynność kory nadnerczy Endokrynologia Polska 2003; 5 (54) 616-622.
12. Oelkers W Adrenal Insufficiency NEJM 1996; 335(16): 1206-1212.
13. Stern N, Tuck ML: The Adrenal Cortex and Mineralocorticoid Hypertension w: Manual of Endocrinology and Metabolism s. 122-128.
14. Laureti S, et al.: Low dose (1 microg) ACTH test in the evaluation of adrenal dysfunction in pre-clinical Addison´s disease. Clin. Endocrinol. (Oxf). 2000; 53: 107-115.
15. Winqvist O, et al.: Two different cytochrome P450 enzymes are the adrenal antigens in autoimmune polyendocrine syndrome type I and Addison´s disease. J. Clin. Invest., 1993; 92: 2377-2385.
16. Coco G, et al.: Estimated risk for developing autoimmune Addison´s disease in patients with adrenal cortex autoantibodies. J. Clin. Endocrinol. Metab., 2006; 91: 1637-1645.
17. Gambelunghe G, et al.: Microsatellite polymorphism of the MHC class I chain-related (MIC-A and MIC-B) genes marks the risk for autoimmune Addison´s disease. J. Clin. Endocrinol. Metab., 1999; 84: 3701-3707.
18. Ghaderi M, et al.: MHC2TA single nucleotide polymorphism and genetic risk for autoimmune adrenal insufficiency. J. Clin. Endocrinol. Metab., 2006; 91: 4107-4111.
19. Arlt W: What´s new in adrenal insufficiency and what´s stress? Materiały dydaktyczne Summer School 13-16 July 2004 St Anne´s College, Oxford University, UK.
20. Russcher H, et al.: Strategies for the Characterization of Disorders In Cortisol Sensitivity. J. Clin. Endocrinol. Metab., 2006; 91: 694-701.
21. van Rossum E and Lamberts S: Polymorphisms in the Glucocorticoid Receptor Gene and Their Associations with Metabolic Parameters and Body Composition Recent Progress in Hormone Research 2004; 59: 333-357.
22. Monson JP: The assessment of glucocorticoid replacement therapy Clinical Endocrinology 1997; 46: 269-270.
23. Crown A, Lightman S: Why is management of glucocorticoid deficiency still controversial: a reviev of the literature. Clinical Endocrinology 2005; 63: 483-492.
24. King JA, et al.: Long-term corticosteroid replacement and bone mineral density in adult women with classical congenital adrenal hyperplasia”. J. Clin. Endocrinol. Metab., 2006; 91: 865-869.
25. Papierska L, Rabijewski M: Osteoporoza posteroidowa Polskie Archiwum medycyny wewnętrznej 2007; 117 (8) In Press (artykuł dostępny online).
26. Dunne F, et al.: Cardiovascular function and glucocorticoid replacement in patients with hypopituitarism. Clinical Endocrinology, 1995; 43, 623-629.
27. Wai Lang Chu: Phoqus completes Phase I Chronocort clinical study www.drugresearcher.com/marketreport.
28. Oelkers W, Lage M: Control of mineralocorticoid substitution in Addison´s disease by plasma renin measurement. Klin Wochenschr 1976; 54: 607-612.
29. Flad TM, et al.: The role of plasma renin activity in evaluating the adequacy of mineralocorticoid replacement in primary adrenal insufficiency. Clin. Endocrinol. (Oxf). 1996; 45(5): 529-34.
30. Arlt W, et al.: Dehydroepiandrosterone Replacement in Women with Adrenal Insufficiency N. Engl. J. Med., 1999; 341: 1013-2.
31. Arlt W: Quality of life in Addison´s disease – the case for DHEA replacement Clinical Endocrinology 2002; 56: 571-7.
