© Borgis - Postępy Nauk Medycznych 3-4/2001, s. 18-20
Witold Palasik
Homocysteina – czynnik ryzyka występowania niedokrwiennego udaru mózgu
Homocysteine – risk factor for ischemic stroke
Klinika Neurologii i Epileptologii Centrum Medyczne Kształcenia Podyplomowego w Warszawie
Kierownik Kliniki: dr hab. med. Urszula Fiszer, prof. nadzw. CMKP
Streszczenie
Podwyższony poziom homocysteiny jest istotnym, niezależnym czynnikiem ryzyka dla chorób układu krążenia w tym również udaru mózgu. Rola homocysteiny w patomechanizmie procesu miażdżycowego i zaburzeń krzepnięcia jest złożona i zależna od wielu czynników takich jak na przykład poziom kwasu foliowego lub witamin B12 i B6. Poza tym stopień uszkodzenia naczyń jest zależny od poziomu homocysteiny w surowicy krwi. Wydaje się, że rozpowszechnienie oznaczania poziomu homocysteiny w surowicy krwi w klinice chorób naczyniowych może odgrywać ważną rolę w profilaktyce i zapobieganiu udarom mózgu.
Summary
A high level of total homocysteine is an strong and independent risk factor for cardiovascular diseases. There is also a significant predictor of cerebrovascular diseases especially of stroke. The role of hyperhomocysteinemia in atherosclerotic and thromboembolic process is very complicated and depended on many other factors like level of foliate, vitamin B12 and B6. There is also important the level of increased serum homocysteine which correlate with state of vascular damage. A widely used examination of this factor may have a benefit role in prevention and treatment of stroke.

Udary niedokrwienne mózgu należą obecnie do najpoważniejszych chorób współczesnego społeczeństwa. Poznanie ich patomechanizmu i czynników ryzyka jest jednym z najważniejszych celów współczesnej neurologii. Tylko poprzez zrozumienie procesów prowadzących do wystąpienia udaru niedokrwiennego możemy doprowadzić do osiągnięcia skutecznych metod zapobiegania lub bardziej efektywnego leczenia. Do najistotniejszych czynników ryzyka należą: nadciśnienie tętnicze, choroby mięśnia serca zwłaszcza przebiegające z zaburzeniami rytmu serca, przebyty w przeszłości incydent o charakterze udaru lub przemijającego niedokrwienia, cukrzyca i wiele innych. W miarę rozwoju nowych metod mamy coraz większe możliwości wnikania in vivo w molekularne podstawy procesów zachodzących w żywym organizmie. Analiza zjawisk immunologicznych zachodzących w czasie udaru mózgu pozwoliła na ustalenie bezpośredniego, bardzo niekorzystnego wpływu zakażenia na wystąpienie udaru. Poznajemy coraz więcej czynników wywołujących proces chorobowy prowadzący w końcowym etapie do wystąpienia niedokrwiennego udaru mózgu.
Postęp badań nad czynnikami nasilającymi proces miażdżycowy w organizmie człowieka, wyniki badań statystycznych, unowocześnienie badań laboratoryjnych zwiększających możliwości określania poziomów różnych substancji powstających w wyniku zaburzeń szlaków metabolicznych stanowią istotny element w poznawaniu przebiegu chorób. Jednym z efektów tych badań jest stwierdzenie zwiększenia poziomu homocysteiny wśród chorych z udokumentowaną chorobą wieńcową, u których przebieg kliniczny był szczególnie niekorzystny. W latach 60. McCully po raz pierwszy doniósł o dwóch przypadkach dzieci z homocystinurią, u których stwierdził znacznie zaawansowane zmiany miażdżycowe w obrębie naczyń tętniczych (1). Postawiona przez niego hipoteza o ewentualnej roli homocysteiny w procesie miażdżycy do początku lat 80-tych była mocno krytykowana.
Homocysteina jest aminokwasem powstającym na szlaku przemian metionina – cysteina. W procesie prawidłowej przemiany bardzo ważną rolę odgrywają między innymi witamina B6 (w procesie transsulfuracji powodującej powstanie cystotioniny), witamina B12 oraz kwas foliowy (w procesie remetylacji). Najbardziej znane defekty przemiany w tym szlaku powstają na dwóch etapach. Pierwszy przy wrodzonym niedoborze beta syntetazy cystotioninowej katalizującej syntezę cystotioniny z homocysteiny i seryny. Drugi defekt, częściej występujący i lepiej poznany, jest spowodowany mutacją w obrębie genu C677T (2) to niedobór enzymu, reduktazy metylenotetrahydrofolianowej, uczestniczącego w remetylacji homocysteiny w metioninę. Efektem zaburzeń tych szlaków metabolicznych jest znaczny wzrost poziomu homocysteiny. Defekt genetyczny ujawnia się bardzo szybko, jeszcze w wieku wczesnodziecięcym. W obrazie klinicznym stwierdzane są zaburzenia rozwoju umysłowego oraz szczególnie nasilony proces miażdżycowy, zupełnie nietypowy dla wieku, mogący powodować zawały serca lub udary niedokrwienne. Obserwowany u tych chorych proces miażdżycowy jest uogólniony i ma typowy obraz histopatologiczny. Zmiany są obserwowane we wszystkich naczyniach zarówno w obrębie ośrodkowego układu nerwowego jak i w naczyniach wieńcowych i innych. Dodatkowo procesowi patologicznemu towarzyszą zaburzenia procesu krzepnięcia. Wada ta dziedzicząca się w sposób autosomalny recesywny ujawnia się klinicznie u homozygot. Zauważono poza tym, że osoby u których stwierdzana jest heterozygotyczna postać zaburzeń przemiany homocysteiny (nie ujawniają się one klinicznie i mogą być wykrywane przypadkowo lub wśród rodzin obciążonych tą wadą przy pomocy specjalnych testów diagnostycznych) obserwuje się znamiennie wczesne występowanie procesu miażdżycowego. Dodatkowo stwierdzono, że nasilenie procesu miażdżycowego jest zależne od poziomu homocysteiny w surowicy krwi (3). Ten szczególny przebieg schorzenia zwrócił uwagę badaczy na homocysteinę jako czynnik indukujący proces miażdżycowy.
Aktualne wyniki badań wskazują na to, że homocysteina może odgrywać rolę w zapoczątkowaniu tego procesu patologicznego. Aminokwas ten ma właściwości cytotoksyczne, które powodują uszkadzanie komórek śródbłonka i prowadzi do nasilenia degradacji elastyny w błonie wewnętrznej; przyspiesza to procesy włóknienia i wapnienia. Dodatkowo uszkadzanie komórek śródbłonka powoduje zaburzenia procesu przylegania płytek. Zjawisko to jest proporcjonalne do stężenia homocysteiny. Kolejnym czynnikiem wzmagającym patologiczne procesy w obrębie naczyń, będących wynikiem wzrostu poziomu homocysteiny w surowicy krwi jest zaburzenie równowagi procesów krzepnięcia. Chorym z hiperhomocysteinemią towarzyszą obniżenia poziomów antytrombiny III, czynnika VII oraz białka C. W miarę analizy kolejnych wyników badań dodatkowo znajdujemy wpływ homocysteiny na coraz bardziej złożone mechanizmy układu krzepnięcia (4, 5) np. poprzez wpływ na działanie tkankowego aktywatora plazminogenu. Jednocześnie homocysteina wykazuje zdolności do tworzenia związków z tlenkiem azotu (NO) powodując pośrednio nasilenie zdolności płytek do agregacji. Homocysteina posiada również zdolności do modyfikacji aktywności lipoproteiny LDL. Doprowadza to do wzrostu cytotoksyczności tej cząsteczki wobec komórek śródbłonka oraz wytwarzania komórek piankowatych, czego efektem jest przyspieszenie procesu miażdżycowego poprzez uszkodzenie ścian naczyń (ryc. 1).
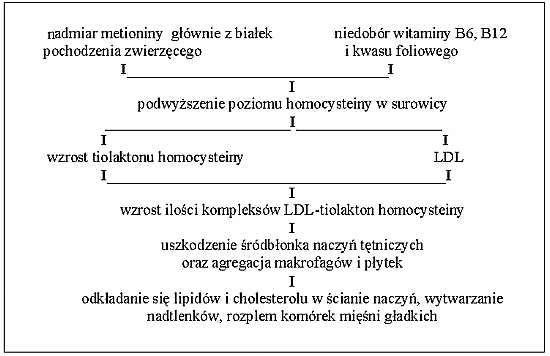
Ryc. 1. Proponowany schemat zmian w przebiegu nadmiaru homocysteiny (wg Larkin, 1998).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Larkin M.: Lancet. 1998, 352 (9137):1364-1368.
2. Morita H. et al.: Arteriosclerosis, Thrombosis & Vascular Biology. 1998, 18(9):1465-9.
3. van den Berg M. et al.: Arterioscler Thromb. Vasc. Biol. 1996, 16: 165-171
4. Lindgren A. et al.: Stroke. 1996, 7(6): 1066-71,
5. Sacco R.L. et al.: Neuroepidemiology. 1998, 17(4):167-73.
6. Selhub J.et al.: Journal of Nutrition. 1996, 126(4 Suppl): 1258S-65S.
7. Selhub J. et al.: New England Journal of Medicine. 1995, 332(5):286-91.
8. Selhub J. Jacques P.F. JAMA1993, 270:2693-8,
9. Rimm E.B. et al.: JAMA, 1998, 279:359-64,
10. Kittner S.J. et al.: Stroke, 1999, 30:1554-60
11. Giles W.H. et al.: Ethnicity & Disease. 1998, 8(2):149-57.
12. Perry I.J. et al.: Lancet. 1995, 346(8987):1395-8.
13. Naruszewicz M. Polskie Archiwum Medycyny Wewnętrznej. 1997 97 Spec No:37-45.
14. Ray J.G. Harvey D.T. Archives of Physical Medicine & Rehabilitation. 1998 79(3):343-5.
15. Hoogeveen E.K. et al.: Arteriosclerosis, Thrombosis & Vascular Biology. 1998 18(1):133-8.
16. Malinow M.R. et al.: Circulation 1993, 87:1107-1110.