© Borgis - Postępy Nauk Medycznych 1-2/2003, s. 40-44
Leszek Pączek, Bartosz Foroncewicz
Tolerancja immunologiczna – wiodącym problemem transplantologii XXI wieku
The immune tolerance – leading problem in transplantology of the XXI century
Klinika Immunologii, Transplantologii i Chorób Wewnętrznych, Instytut Transplantologii Akademii Medycznej w Warszawie
Kierownik Kliniki: prof. dr hab. med. Leszek Pączek
Streszczenie
Definicją tolerancji jest brak odpowiedzi immunologicznej na antygeny allograftu bez potrzeby stosowania przewlekłego leczenia immunosupresyjnego. Według tej definicji tolerancja jest największym wyzwaniem medycyny transplantacyjnej XXI wieku. Jakkolwiek wiele badań na zwierzętach udowodniło, że wywołanie tolerancji immunologicznej jest możliwe, u ludzi indukcja tolerancji nadal postrzegana jest bardziej jako teoria niż praktyka.
Summary
The tolerance means lack of immune response to allograft antigens without a need of chronic immunosuppressive therapy. According to such definition, the immune tolerance remains one of the biggest challenges of the transplantology in XXI century. Although many experiments on animals proved that it is possible to induce tolerance, the experience in human remains far from satisfactory.

WPROWADZENIE
Swoista tolerancja jest to stan, w którym układ odpornościowy nie rozwija odpowiedzi przeciwko danemu antygenowi (lub grupie antygenów) ale odpowiedź skierowana przeciwko pozostałym antygenom jest utrzymana. Naukowa koncepcja tolerancji immunologicznej została opracowana przez Burnetta i Billinghama przed 50 laty. Ogłoszona wówczas teoria zakładała, że w życiu płodowym człowiek „uczy się” odróżniać własne tkanki, co pozwala na identyfikację zewnętrznych czynników infekcyjnych w okresie po urodzeniu.
Centralnie tolerancja zachodzi głównie w grasicy oraz częściowo w szpiku kostnym. Ponad 95% limfocytów T ulega destrukcji na drodze apoptozy. Sygnałem wyzwalającym jest pozytywna reakcja limfocyta T na antygeny własne, prezentowane przez układ MHC (Major Histocompatibility Complex). Limfocyt T, którego receptor TcR (T cell receptor) ma niskie powinowactwo lub brak powinowactwa do prezentowanych własnych antygenów, uzyskuje dodatnią regulację genu Bcl-2 i unika śmierci na drodze apoptozy. Proces ten określany jest mianem pozytywnej selekcji klonów. Warto w tym miejscu przypomnieć, że gen Bcl-2 jest indukowany przez leki immunosupresyjne z grupy inhibitorów mTOR (mammalian target of rapamycin).
W szpiku kostnym zachodzi podobny proces, ale w odniesieniu do limfocytów pre-B. Dochodzi do apoptozy limfocytów B (IgM+, IgG-). Limfocyty reagujące na antygeny własne są usuwane z krążenia i niszczone. Proces usuwania autoreaktywnych limfocytów B jest jednak mniej precyzyjny. Okazało się, że zmiana łańcucha lekkiego (tzw. receptor editing) dla receptora BcR (B cell receptor) pozwala uniknąć apoptozy. Jest to potencjalny mechanizm wymykania się autoreaktywnych limfocytów B spod opisywanej powyżej kontroli.
Główne białka hamujące apoptozę należą do grupy 15 białek określanych mianem Bcl. Nazwa pochodzi od B-cell lymphomas, gdzie obserwowano nadekspresję tych białek. Białka Bcl wpływają zarówno na błony mitochondrium, jak również bezpośrednio inaktywują kaspazę 9.
Interesującym jest, że dysponujemy w klinice lekami hamującymi Bcl, a tym samym nasilającymi apoptozę. Są to inhibitory mTOR (rapamycyna) (1). Komórki ssaków, w tym układu immunologicznego, zawierają w cytoplazmie białko hamujące proces apoptozy określane jako IAP (intracellular inhibitors of apoptosis) (2). Zidentyfikowano receptory odpowiedzialne za indukcję apoptozy. Są to: Fas i receptor dla TNF (Tumour Necrosis Factor) – TNFR. Dalsze badania wykazały, że również antygeny zgodności tkankowej DR3, DR4, DR5 i DR6 mogą pełnić rolę receptorów śmierci dla komórek. Biologiczne znaczenie aktywacji przez cząstki MHC klasy II (DR) nie jest jasne (3). Należy pamiętać, że zrozumienie mechanizmów regulujących apoptozę nie jest pełne. Również udział apoptozy w indukcji tolerancji nie jest w pełni wyjaśniony. Przykładowo, myszy transgeniczne pozbawione zdolności do produkcji TNFa nie rozwijają chorób autoimmunologicznych, mimo że mają zaburzony system indukowania apoptozy.
Jednym z pierwszych mechanizmów wywoływania tolerancji był układ matka-płód. Przedstawiono ten układ jako anatomiczną sekwestrację płodu poprzez łożysko. Rozumowanie takie jest jednak zbytnio uproszczone. Wynika to z faktu, że tkanki matki i płodu kontaktują się bezpośrednio na obwodzie łożyska. Dodatkowo, krew matki (z limfocytami) opływa kosmki trofoblastu (tkanka płodu). Szczegółowe badania wykazały, że włączone są aktywne mechanizmy czynnościowe. Po stronie trofoblastu (płodu) są to: zmiana typu HLA-A na G, wzmożona ekspresja ligandu Fas-L i TNFa oraz odpowiedź typu Th2. Dodatkowo pojawiają się mechanizmy blokujące odpowiedź nieswoistą indukowaną przez dopełniacz. I tak obserwuje się wzmożoną ekspresję CD46 i CD55 (oba czynniki obniżają aktywność konwertazy C3) oraz CD59 (hamuje aktywność kompleksu C5-9).
Również po stronie matki dochodzi do istotnych zmian w funkcjonowaniu lokalnego układu immunologicznego. Obserwowany jest wzrost aktywności 2,3-dioksygenazy indoloaminy, co prowadzi do deficytu tryptofanu. Limfocyty T cytotoksyczne są szczególnie wrażliwe na brak tego aminokwasu w środowisku. Bardziej istotną zmianą jest jednak istotnie zmniejszona aktywacja receptora dla IL-2 z jednocześnie wysokim poziomem IL-15.
Reasumując, sekwestracja anatomiczna jest zwykle wspomagana przez aktywne mechanizmy czynnościowe. W medycynie transplantacyjnej istnieje kusząca droga sekwestracji wybranych komórek, a nawet tkanek (n.p. wyspy trzustkowe) w półprzepuszczalnych sferach. Materiał zastosowany do takiej procedury również przepuszcza tlen i składniki odżywcze, ale nie powinien przepuszczać przeciwciał i składowych dopełniacza. Opłaszczenie komórek/tkanek nie pozwala na wrastanie naczynia, nie pozwala na przechodzenie białek transportujących hormony, a także jonów żelaza. Dodatkowo, mechanizmy nieswoiste indukują fazę reakcji na ciało obce, co prowadzi do włóknienia wokół opłaszczonych komórek.
MECHANIZMY TOLERANCJI
Uważa się, że inukcja tolerancji może zachodzić w kilku mechanizmach. Są to: wywołanie anergii allospecyficznych limfocytów T poprzez blokadę kostymulacji lub poprzez allopeptydy oderwane od cząstek MHC klasy I; indukcja regulatorowych komórek T (Tr1); przesunięcie odpowiedzi komórek Th1 w kierunku Th2 oraz usunięcie allospecyficznych limfocytów T (poprzez negatywną selekcję w grasicy lub poprzez efekt cytotoksyczny).
Anergia limfocytów T
Komórki prezentujące antygen (APC = Antigen Presenting Cells) rozpoznają obce antygeny, a następnie w kontekście antygenów głównego układu zgodności tkankowej MHC klasy II, poprzez receptor TRC prezentują je limfocytom Th, co prowadzi do fazy efektorowej. Kluczowym zagadnieniem dla transplantologów jest to, czy komórka efektorowa po otrzymaniu informacji, pozostanie anergiczna, czy też zginie w mechanizmie apoptozy. Równocześnie z przekazywaniem informacji do limfocytu T mają miejsce zjawiska tzw. kostymulacji, w których biorą udział m.in. CD80 (B7.1) i CD86 (B7.2) z cząsteczkami CD28 oraz CD40 i CD40L (ryc.1). Jeżeli, z różnych powodów, powyższa kostymulacja limfocyta T nie nastąpi, to pomimo przekazania sygnału nie dojdzie do proliferacji limfocyta T (ryc. 2). Stan taki nazywamy anergią limfocytu T. Leki immunosupresyjne wywierają efekt już wewnątrz limfocytu T, wpływając na zupełnie inne procesy, stąd liczne próby wywołania anergii poprzez blokadę dróg kostymulacji. W układzie doświadczalnym zablokowanie tych dwóch dróg, bez dodatkowej immunosupresji powoduje studniowe przeżycie przeszczepu trzustki u szczurów. Nawet w tak wysoce immunogennym modelu, jak ksenotransplantacja szczurzych wysp trzustkowych myszom, zablokowanie drogi CD28 / B7 rozpuszczalnym białkiem fuzyjnym CTLA-4Fc oraz drogi CD40 / CD40L-monoklonalnym przeciwciałem anty – CD40L wywołało stan tolerancji (4). Jej utrzymanie w tym modelu nie było związane z przesunięciem odpowiedzi Th1 na Th2, lecz z gromadzeniem się w przeszczepionym narządzie dużej liczby nieaktywnych limfocytów T. W takim samym modelu, zablokowanie drogi kostymulacji jedynie białkiem CTLA-4Fc nie wywołało tolerancji (5). Było natomiast skuteczne w innych modelach m.in. na myszach (6, 7) oraz wywoływało anergię limfocytów T in vitro (8). Podawanie szczurom przeciwciał anty- CD40L przez 6 miesięcy wywoływało ponadroczne przeżycie przeszczepionych nerek bez dodatkowej immunosupresji (9).
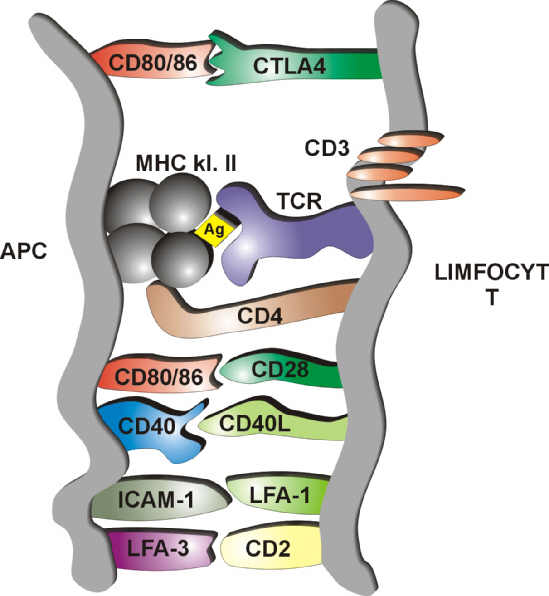
Ryc. 1. Drogi kostymulacji limfocyta T. Objaśnienia w tekście.
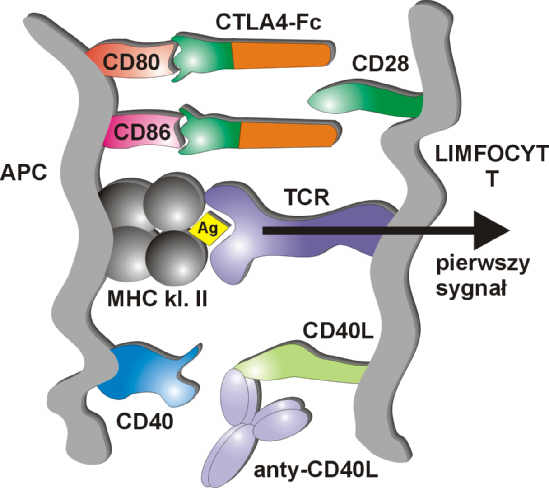
Ryc. 2. Zablokowanie dróg kostymulacji poprzez podanie CTLA-4-Fc i przeciwciał anty-CD40L. Objaśnienia w tekście.
Badając biorców komórek macierzystych szpiku od dawców spokrewnionych z jedynie częściową zgodnością MHC, którym wraz z przeszczepianymi komórkami podawano CTLA4-Fc ex vivo, uzyskano znaczącą, jakkolwiek nie całkowitą redukcję choroby GvH (przeszczep przeciwko gospodarzowi) (10). W innych próbach klinicznych zablokowanie drogi kostymulacji przeciwciałami anty-CD28, w skojarzeniu z monoterapią cyklosporyną, nie tylko zmniejszyło częstość choroby GvH, ale również pozwoliło biorcy zachować zdolność odpowiedzi na antygeny wirusowe i antygeny komórek białaczki (11, 12). Zarówno CTLA-4-Ig, jak też anty-CD40L są oceniane w badaniach I fazy.
Dostarczenie odpowiedniej ilości rozpuszczalnych antygenów MHC klasy I, np. stosując terapię genową może również wywołać anergię limfocytów T (13). Badania na szczurach pokazały, że w przeszczepach wielonarządowych, kiedy biorca styka się ze zwielokrotnioną ilością obcych antygenów dochodzi do indukcji tolerancji, również gdy jednym z przeszczepianych narządów nie jest wątroba, która jak wiadomo jest olbrzymim źródłem antygenów MHC. Udowodniono, że wątroba człowieka produkuje aż połowę krążących antygenów MHC klasy I (14). Osobno lub w połączeniu z przeciwciałami są one po transplantacji istotą zmian zachodzących w układzie immunologicznym, jednak u ludzi przeszczepienie wątroby bez leczenia immunosupresyjnego nie jest w stanie wywołać tolerancji. Udaje się to na modelach zwierzęcych. U szczurzych biorców wątroby wykazujących tolerancję stwierdza się wysoką zawartość cząstek MHC klasy I, wspólnych z dawcą. Wstrzykiwanie surowic lub chłonki tych zwierząt innym (tzw. naiwnym) biorcom wydłuża przeżycie przeszczepionego serca lub skóry (15). Podejmowano próby określenia udziału komórek śródmiąższu wątroby, bogatych w cząstki MHC oraz tzw. komórek pasażerskich, pochodzących ze szpiku w procesie indukcji tolerancji. Komórki pasażerskie, do których należą limfocyty, komórki dendrytyczne i komórki Browicza-Kupfera wykazują destrukcyjną aktywność w chorobie GvH oraz po przeszczepach wątroby, szczególnie u biorców z obniżoną odpornością. Mają zdolność chimeryzmu i zdarza się, że komórki Browicza-Kupfera po kilku tygodniach od przeszczepienia wątroby stają się komórkami gospodarza. Jest to jeden z mechanizmów tłumaczących tolerancję. W jednym z badań na szczepie szczurów, u których uprzednio obserwowano tolerancję po 30 dniach od przeszczepienia wątroby, większość komórek pasażerskich uległa mikrochimeryzmowi, przekształcając się w komórki gospodarza. Wówczas dokonano retransplantacji wątroby kolejnym biorcom, zarówno ze szczepu dawców, jak też biorców. W ten sposób wykazano, że do indukcji tolerancji niezbędne są obydwa mechanizmy (16).
W erze przed stosowaniem cyklosporyny wykazano, że BST (blood specific transfusion) przed operacją przeszczepienia nerki pobranej od spokrewnionego dawcy istotnie poprawia wyniki przeżycia przeszczepu. W ostatnich latach ponownie powrócono do koncepcji immunizacji antygenami HLA (praktycznie stosowane są peptydy homologiczne do łańcuchów a lub b układu HLA). W doświadczeniach na zwierzętach wykazano, że preimmunizacja antygenami HLA indukuje częściową tolerancję (17).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Adams Jm., Cory S: Science 1998, 281: 1322-5.
2. Roy N. et al.: EMBOY 1997; 16: 6914-17.
3. Nagata S: Cell 1997, 58: 355-361.
4. Lehnert A.M. et al.: Transplantation, 2000; 69:1176-1185.
5. Tran H.M. et al.: Xenotransplantation, 1997; 4:222.
6. Pearson TC. et al.: Transplantation, 1994; 57:1701.
7. Sayegh M.H., Nickerson P.W., Hannock W.W. et al.:J Exp Med., 1995;181:1869.
8. Tan P. et al.: J Exp Med., 1993;177:165.
9. Turka L.A. et al.: Graft, 1998;1:111-116.
10. Guinan E.C. et al.: New Engl J Med., 1999; 340:1704.
11. van Gool S.W. et al.: Blood, 1994; 83:176.
12. Comoli P. et al.: Blood, 1997 (suppl.1):535a (abstrakt).
13. Pleyer U. et al.: Immunology Today, 2000;21:12-14.
14. Pollard S. et al.: Transplantation 1990; 49:659-660.
15. Roser B.J. et al.: Liver Transplantation; London: Grune & Stratton; 1987:35-54.
16. Srivatanawongsa V. et al.: Nat Med., 1995;1:428-432.
17. Murphy B., Krensky A.M.: J Am Soc Nephrol 1999, 10: 1346-1355.
18. Montagna D. et al.: Blood 1999; 93:550.
19. Cavazzana-Calvo M. et al.: Blood 1994; 83:288.
20. Valteau-Couanet D. et al.: Transplantation, 1993; 56:1574.
21. Larsen C. et al.: Nature 1996;381:434-438.
22. Onodera K. et al.: J Immunol, 1998; 160:5765-5772.
23. Graca l., Honey K., Adams E. et al.: The J of Immunol 2000; 165:4783-4786.
24. Ciubotariu R. et al.: Human Immunology 2001, 62: 15-20.
25. Cortesini R. et al.: Immunol Rev 2001, 182: 201-206.
26. Li X. et al.: Current Opinion in Immunology 2000, 12: 522-27.
27. Bromberg J.S.: Adv Immunol, 1998; 69:353-409.
28. Thompson C.B. et al.: Immunity, 1997; 7:445-450
29. Calne R.Y. et al.: Lancet, 1998; 351:1701-1702.
30. Calne R.Y. et al.: Transplantation, 1999; 68:1613-1616.
