© Borgis - Postępy Nauk Medycznych 10/2010, s. 784-793
*Miłosz Parczewski
Genetyczna oporność na zakażenie HIV
Genetic resistance to HIV infection
Katedra i Klinika Chorób Zakaźnych i Hepatologii Pomorskiej Akademii Medycznej w Szczecinie
Kierownik Kliniki: prof. dr hab. med. Anna Boroń-Kaczmarska
Streszczenie
Warianty genetyczne związane z opornością na zakażenie HIV dotyczą zmienności w układzie HLA, wariantów genów receptorów cytokin i chemokin oraz ich ligandów. Warianty receptorów chemokin i ich ligandów powodują zmiany w ilości receptora na powierzchni komórek, a co za tym idzie mogą zmniejszać lub zwiększać ilość dostępnych „bram”, przez które HIV wnika do komórki, modyfikując podatność na zakażenie oraz wpływając na szybkość progresji. Dodatkowo na zakażenie podatnych populacji komórkowych wpływa zmienność genetyczna regulująca stężenie ligandu dla danego koreceptora (polimorfizmy RANTES, SDF, IL4i IL10), które mogą współzawodniczyć z wirusem o dany receptor lub powodować zmiany w gęstości koreceptora na powierzchni komórkowej w mechanizmie internalizacji. Polimorfizmy receptorów chemokin i ich ligandów nie są jedynym czynnikiem wpływającym na zakażenie HIV, ale wraz z polimorfizmami w układzie HLA, zmiennością genetyczną samego wirusa tworzą skomplikowany układ wielu zmiennych opisanych w niniejszej pracy.
Summary
Variants conferring genetic resistance to HIV infection are observed within the HLA system, chemokine and cytokine receptor genes but also their ligands. Genetic variants of chemokine receptors play a role in modification of the HIV coreceptor availability on the cellular surface and thus influence susceptibility to infection and its progression. Additionally infection pace is related to the ligand expression – therefore regulatory variants in this system might result in overproduction of chemokines, coreceptor internalization and competitive inhibition of viral entry into the cell ( RANTES, SDF, IL4i IL10 polymorphisms). All above mentioned factors influence HIV progression and interplay in the multilayer system described in this review study.

Wprowadzenie
Zakażenie ludzkim wirusem niedoboru odporności typu 1 (HIV-1), a często także zdolność do kontroli tego zakażenia jest regulowana przez równowagę przynajmniej trzech czynników: zdolności gospodarza do wytwarzania skutecznej odpowiedzi immunologicznej, sekwencji RNA wirusa oraz indywidualnej genetycznej podatności/oporności gospodarza.
Klinicznie dwa zjawiska dowodzą istnienia naturalnej oporności na zakażenie HIV-1. Pierwsze jest związane z przypadkami osób, które pozostały niezakażone mimo (często wielokrotnej) ekspozycji na HIV-1 ( highly exposed persistently HIV seronegative). Obserwacje zjawiska były związane z kobietami trudniących się prostytucją w Nairobi (1), ale dotyczyły one również partnerów seksualnych osób zakażonych HIV, transmisji wertykalnych, używania narkotyków dożylnych czy ekspozycji zawodowych.
Drugie to zjawisko braku progresji choroby mimo zakażenia ( long term nonprogression) definiowane jako utrzymujący się niski poziom wiremii HIV i wysokiej liczby limfocytów CD4+ w surowicy oraz brakiem klinicznych cech progresji do AIDS. W piśmiennictwie nie ma jednoznacznych kryteriów, po jakim czasie można uznać, że u danej osoby obserwuje się opisane wyżej zjawisko – okres wynosi od 8-15 lat (2-5). Zjawiskiem odwrotnym jest szybka progresja ( rapid progression) – w takim przypadku kliniczne wystąpienie AIDS obserwuje się po 3 latach (lub szybciej) od zakażenia (4, 5).
Obecnie sugeruje się, że oba zjawiska są ze sobą ściśle związane – czynniki, które chronią lub ograniczają wnikanie HIV do podatnych populacji komórkowych będą z jednej strony zmniejszały podatność na zakażenie, a z drugiej spowalniały lub całkowicie eliminowały postęp choroby (6).
Warianty związane z opornością na zakażenie HIV
Zakładając, że receptory chemokin są „bramami” dla wirusa HIV umożliwiającymi mu zakażenie komórek prezentujących na powierzchni koreceptor, powiedzenie Hannibal ante portas! byłoby dobrą przenośnią. Badania epidemiologiczne prowadzone na grupach osób wielokrotnie eksponowanych na HIV, które nie uległy zakażeniu, sugerowały istnienie zjawiska naturalnej oporności na zakażenie. Obserwacje prowadzone na grupach o zróżnicowanym sposobie narażenia – użytkowników narkomanii dożylnej, osób eksponowanych drogą seksualną, dzieci matek zakażonych HIV – opisywały istnienie naturalnej, choć rzadkiej, genetycznej oporności na HIV, która różniła się w zależności od tła etnicznego (6-8). Odkrycie, że wirus potrzebuje koreceptora pod postacią receptora dla chemokin oraz tego, że to właśnie warianty genetyczne związane z kodowaniem receptorów chemokin i ich ligandów są jednym z czynników związanym z modyfikacją podatności na zakażenie i jego progresją, spowodowało, że pozostają one w centrum zainteresowania badawczego (9-18).
W początkowej fazie zakażenia HIV łączy się preferencyjnie z CCR5, dominują szczepy makrofagotropowe R5, nie tworzące syncytiów w hodowlach komórkowych. Zmiana tropizmu wirusa następuje typowo po kilku latach (około 5) z pojawieniem się najpierw wirusów o podwójnym tropizmie R5X4, a następnie dominacją limfotropowych szczepów X4, związanych z zaawansowanym zakażeniem.
Warianty receptorów chemokin i ich ligandów powodują zmiany w ilości receptora na powierzchni komórek, a co za tym idzie mogą zmniejszać lub zwiększać ilość dostępnych „bram”, przez które HIV może wnikać do komórki i w tym mechanizmie modyfikować podatność na zakażenie oraz wpływać na szybkość progresji. Dodatkowo na zakażenie podatnych populacji komórkowych wpływa zmienność genetyczna regulująca stężenie ligandu dla danego receptora (polimorfizmy RANTES, SDF, IL4i IL10 (19-25), które mogą współzawodniczyć z wirusem o dany receptor lub powodować zmiany w gęstości koreceptora na powierzchni komórkowej w mechanizmie internalizacji. W tabeli 1 przedstawiono wybrane warianty genetyczne receptorów chemokin i ich ligandów wraz z danymi z piśmiennictwa dotyczącymi ich wpływu na ryzyko zakażenia HIV i progresję do AIDS.
Tabela 1. Polimorfizmy układu receptor chemokin – ligand a ich wpływ na zakażenie HIV-1 i progresję do AIDS.
| Wariant genu | Efekt | Wpływ na ryzyko zakażenia HIV-1 | Wpływ na progresję zakażenia HIV-1 |
| Polimorfizmy ligandów dla CCR5 i CXCR4 |
| Polimorfizm promotora RANTES kodon (-28) allel G | Zwiększenie aktywności promotora | Brak danych w piśmiennictwie | Spowalnia (21) |
| Polimorfizm promotora RANTES kodon (-403) allel A | Zwiększenie aktywności promotora | Zmniejsza (19) | Spowalnia (19) |
| Polimorfizm SDF1 w nie ulegającym transkrypcji regionie 3', kodon 801 allel A | Zmiana sposobu składania mRNA - alternative splicing | Brak danych w piśmiennictwie | Spowalnia (22, 26, 27) |
| Polimorfizmy CCR5 |
| Delecja 32 par zasad CCR5 | Utrata dwóch domen przezbłonowych receptora | Zmniejsza (14, 17, 28, 29) | Spowalnia (14, 28, 30) |
| Tranzycja T/A w części kodującej CCR5, kodon 303 | Powstanie sygnału terminującego translację ("stop kodon") i synteza niefunkcjonalnego białka | Zmniejsza (31) | Spowalnia (31) |
| Polimorfizm promotora CCR5 pozycja (-2459) allel G | Regulacja efektywności transkrypcji CCR5 | Sprzeczne dane w literaturze:
- Zwiększa (32)
- Zmniejsza (33) | Spowalnia (34) |
| Polimorfizm promotora CCR5 pozycja (-2132) allel T | Zmniejsza (35) | |
| Polimorfizm promotora CCR5 pozycja (-2086) allel G | Zmniejsza (2) | |
| Polimorfizmy innych receptorów chemokin |
| CCR2 pozycja 190 allel A | Niewyjaśniony - prawdopodobnie modulacja funkcji CCR5 (100) | Doniesienia niejednoznaczne:
- nie wpływa (37-39)
- zmniejsza (32) | Spowalnia (38, 40) |
| CX3CR1 744 allel A i pozycja 838 allel G | Zmniejszenie ilości funkcjonalnego receptora na powierzchni komórek | Brak danych w piśmiennictwie | Sprzeczne dane w literaturze:
Przyspiesza (15)
Spowalnia (42) |
Polimorfizmy receptorów chemokin i ich ligandów nie są jedynym czynnikiem wpływającym na zakażenie HIV, ale wraz z polimorfizmami w układzie HLA i zmiennością genetyczną samego wirusa tworzą skomplikowany układ wielu zmiennych. Z dużym prawdopodobieństwem można założyć, że większość czynników uwarunkowanych genetycznie, a modyfikujących progresję choroby, będzie również wpływała na podatność na zakażenie wirusem. Taka hipoteza została wysunięta przez Marmor i wsp. po analizie badań związanych z podatnością na zakażenie i progresję. Jest to „teoria unifikacji” sugerująca, że zmienność genetyczna „gospodarza”, która chroni lub utrudnia wniknięcie HIV-1 do komórki, będzie nie tylko zmniejszała podatność na zakażenie, ale również spowalniała lub całkowicie eliminowała rozwój AIDS (6).
Układ HLA
Allele HLA są kodowane przez liczne loci, leżące na chromosomie 6p21.31, pozostającym najbardziej zmiennym regionem ludzkiego genomu. Zarówno w przypadku HLA I, jak i HLA II, następuje łączenie się z określonymi peptydami lub receptorami komórek T ( T-cell receptor). Zmienność polimorficzna HLA jest między innymi uzależniona od presji selekcyjnej wywieranej przez czynniki infekcyjne, z potencjalną korzyścią w przypadku heterozygot (43). Pozwala ona na łączenie i prezentację większej ilości peptydów. Dlatego duża ilość alleli ma funkcjonalne znaczenie dla podatności oraz odpowiedzi immunologicznej na patogeny. Należy podkreślić, że między populacjami istnieje znacząca zmienność częstości występowania poszczególnych alleli, najprawdopodobniej związana z działaniem zróżnicowanych presji selekcyjnych na te populacje w przeszłości. MHC I i II odgrywają pierwszoplanową rolę w regulacji odpowiedzi immunologicznej w przypadku zakażeń wirusowych, a co za tym idzie w przypadku zakażenia HIV.
Udowodniono, że kontrola spadku poziomu limfocytów CD4 w surowicy przez cytotoksyczne limfocyty T jest procesem immunologicznym zarówno chroniącym przed zakażeniem HIV, jak również ograniczającym postęp choroby w przypadku skutecznego zakażenia (43, 44). Zmienność alleliczna powoduje zmienną siłę wiązania antygenów i wpływa na ich prezentację oraz skuteczne rozpoznawanie przez limfocyty T, a co za tym idzie na jakość odpowiedzi immunologicznej. Presja środowiskowa wywierana przez zróżnicowane allele HLA na replikację HIV, ulegający szybkim mutacjom, może stymulować namnażanie wirusów o skróconej lub wydłużonej żywotności, modyfikując jednocześnie przebieg zakażenia.
Polimorfizmy HLA są związane z ochroną przed zakażeniem HIV, zwiększoną podatnością na zakażenie HIV oraz przyspieszeniem lub opóźnieniem progresji choroby w przypadku osób zakażonych. Dotychczasowe liczne badania zostały przeprowadzone na różnych populacjach i rasach (wymieniając tylko najbardziej istotne: Afryka – Nairobi, Kenia, Gambia; Azja – Tajlandia; Ameryka Północna i Południowa – Argentyna, Stany Zjednoczone Ameryki; Australia), w grupach prezentujących zróżnicowane zachowania ryzykowne (prostytucja, transmisje okołoporodowe, biorcy krwi, kontakty homoseksualne między mężczynami, używanie narkotyków dożylnych) (43-47). Allele HLA związane z mniejszą podatnością na zakażenie HIV to między innymi HLA II DRB1*01 i supergenotyp A2-A*6802 (badanie wykonane na populacji prostytutek z Nairobi) (46), HLA B35 (badanie eksponowanych niezakażonych HIV prostytutek w Gambii) (8), DRB1*1501 i DRB1*13 (*1301-3) (ochrona przed transmisją perinatalną) (47). Natomiast wariant HLA B*5701 wpływa na znaczące wydłużenie czasu bezobjawowego zakażenia u osób żyjących z HIV (48).
Polimorfizmy genów cytokin
Dotychczasowe badania dotyczące związku między cytokinami, allelami modyfikującymi poziom syntezy tych białek a podatnością na zakażenie HIV i jego progresją nie są tak obszerne jak w przypadku HLA czy chemokin, a wyniki tych badań są często sprzeczne. Dotychczas związano podatność na zakażenie HIV lub wpływ na progresję zakażenia z wariantami genów dwóch interleukin – IL4i IL10.
Allel (-589)T promotora genu interleukiny 4 wywiera ochronny wpływ na transmisję drogą heteroseksualną (badania wykonane na populacji japońskiej) poprzez zmniejszanie ilości CCR5 na powierzchni komórki (23). Natomiast w przypadku osób zakażonych promuje on selekcję szczepów X4 i przyspiesza progresję choroby. Początkowe doniesienia, że allel T(-589) IL4spowalnia progresję choroby u osób już zakażonych, nie zostały potwierdzone (49).
Drugą cytokiną, która wpływa na replikację HIV-1, jest interleukina 10. Polimorfizm pojedynczego nukleotydu w promotorze genu IL-10 (substytucja cytozyny na adeninę w pozycji -592) jest związany ze zmniejszeniem syntezy IL-10, co może prowadzić do przyspieszenia namnażania się wirusa, szybszego pojawienia się szczepów T-tropowych i zwiększeniem ryzyka wystąpienia AIDS, co potwierdzono w badaniach przeprowadzonych przez Shin i wsp (24). Aktualnie brak jest danych w piśmiennictwie wiążących polimorfizm IL-10A(-592) z podatnością na zakażenie HIV.
Warianty sekwencji genów receptorów chemokin
Zdolność HIV do wnikania do wnętrza komórki jest bezpośrednio zależna od wiązania z koreceptorem. Dlatego kolejnym istotnym czynnikiem modulującym zarówno podatność na zakażenie, jak też progresję do AIDS, są warianty genetyczne związane z receptorami chemokin i ich ligandami (20).
Chemokiny są małymi białkami (8-12 kilodaltonów) kontrolującymi migrację komórkową, produkowanymi lokalnie w tkankach i działającymi na leukocyty poprzez specyficzne receptory. Pozostają wielofunkcyjną grupą cytokin o właściwościach chemotaktycznych ( CHEMOtactic cytoKINES). Funkcjonalnie można je podzielić na dwie kategorie. Do pierwszej należą te, których sekrecja ma charakter stały, konstytutywny, zaangażowane są w regulację procesów hemostatycznych, związanych z kontrolą migracji limfocytów i nadzorem immunologicznym. Do drugiej chemokiny – produkowane przez komórki po bodźcu zapalnym, które kierują migracją w kierunku miejsca infekcji lub uszkodzenia (50-52).
Chemokiny są substancjami strukturalnie pokrewnymi, zawierającymi cztery cysteiny ułożone w charakterystycznym stałym układzie. W zależności od ułożenia dwóch końcowych cystein mogą być one podzielone na podrodziny: CXC – to a chemokiny, w przypadku których cysteiny są oddzielone pojedynczym aminokwasem, CC – β chemokiny – z cysteinami ułożonymi obok siebie. Istnieją dwie cząsteczki zaliczone do rodziny chemokin, które posiadają tylko dwie zamiast czterech cystein. Są to: limfotaktyna zaliczona do podrodziny γ, która mimo podobnej wielkości i struktury zawiera tylko jedną cysteinę na N-końcu, oraz δ chemokina o strukturze CX3C z trzema aminokwasami między dwiema pierwszymi cysteinami, a w swej strukturze posiadająca szypułę mucynową – fraktalkina (CX3CL1) (53, 54).
W patogenezie zakażenia HIV bezpośrednią kluczową rolę grają chemokiny będące ligandami receptorów, używanymi przez wirus do integracji z komórką CD4+. Ligandy dla receptorów głównych CCR5 i CXCR4 dla wybranych koreceptorów „alternatywnych” – używanych przez niektóre szczepy wirusa – zostały przedstawione w tabeli 2.
Tabela 2. Receptory chemokin związane z patogenezą zakażenia HIV i ich ligandy.
| Receptor | Ligandy | Receptor | Ligandy |
| CCR5 | CCL4 (MIP-1b)
CCL3 (MIP-1a)
CCL5 (RANTES)
CCL2 (MCP-1)
CCL8 (MCP-2)
CCL7 (MCP-3)
CCL13 (MCP-4)
CL11 (MCP-5
- eotaksyna) | CCR3 | CCL11 (MCP-5 - eotaksyna)
CCL7 (MCP-3)
CCL13 (MCP-4)
CCL24 (MPIF-2 - eotaksyna-2)
CCL26 (MIP 4a - eotaksyna-3)
CCL5 (RANTES)
CCL15 (MIP-5 - leukotaktyna-1) |
| CXCR4 | CXCL12 (SDF-1) | CCR8 | CCL-17 (TARC)
CCL-4 (MIP-1B) |
| CCR2 | CCL2 (MCP-1)
CCL8 (MCP-2)
CCL7 (MCP-3)
CCL13 (MCP-4)
CCL11 (MCP-5 - eotaksyna) | CX3CR1 | CX3CL1 (fraktalkina) |
Naturalne ligandy powodują zmniejszenie ilości receptora na powierzchni komórki, mogą również hamować wiązanie wirusa na zasadzie współzawodnictwa (55, 56). Farmakologiczne substancje będące sztucznymi ligandami receptorów chemokin stanowią jeden z istotnych kierunków rozwoju terapii. Przykład mogą stanowić zmodyfikowane analogi RANTES hamujące wiązanie HIV z komórkami Langerhansa, peptydy blokujące część receptora lub zmieniające jego konformację, czy pochodne SDF-1 (40, 57, 58).
Zmienność genetyczna związana z układem receptor chemokinowy – ligand i opornością na zakażenie HIV może dotyczyć zwiększenia stężenia chemokiny (ligandu) w surowicy, skutkując z jednej strony współzawodnictwem z wirusem o wiązanie z receptorem, a z drugiej zmniejszeniem ilości dostępnego receptora na powierzchni komórki (internalizacja), modyfikacji ilości dostępnych koreceptorów na powierzchni komórki w wyniku zmiany aktywności części promotorowej genu kodującego dany koreceptor czy też mutacji w części kodującej genu i w efekcie syntezy niefunkcjonalnego białka, co samo w sobie uniemożliwia połączenie wirusa z komórką (55).
Innym rodzajem zmienności genetycznej, która może ograniczać fuzję wirusa z komórką i podatność na zakażenie oraz spowalniać jego progresję, jest tworzenie dimerów między koreceptorami. Ponadto mutacje związane z modyfikacją przebiegu choroby mogą dotyczyć innych koreceptorów chemokin, które nie wpływają bezpośrednio na stężenie koreceptora głównego, ale modyfikują przebieg zakażenia niektórych subpopulacji komórkowych (59).
W przypadku CCR5 genetycznie uwarunkowana zmiana podatności na zakażenie może być związana z trzema mechanizmami:
a) zróżnicowaną aktywnością promotora modulującego gęstość receptora na powierzchni komórki związaną z jego wariantami (33, 45, 60),
b) mutacjami w części kodującej (delecja 32 par nukleotydów (Δ32), mutacja T/A w pozycji 303) (6, 20, 61),
c) wariantem A190 CCR2, który stabilizuje cząsteczkę CCR2i prawdopodobnie zmniejsza ekspresję CCR5 poprzez wpływ na proces jego dojrzewania w cytoplazmie (22, 36, 37, 62).
Część kodująca genu CCR5
W 1996 roku pojawiły się pierwsze doniesienia na temat oporności na zakażenie u osób ze zmutowanym allelem genu CCR5– delecją 32 par nukleotydów w jego części kodującej, powodującej zmianę struktury części przezbłonowej receptora – utratę trzech z siedmiu domen (ryc. 1a i 1b) (7, 11, 14). Mutacja, nie powodując znaczących defektów immunologicznych, poza zwiększoną podatnością na objawowe zakażenie Wirusem Zachodniego Nilu (63), jest związana z ograniczeniem integracji HIV z komórką poprzez zmniejszenie ilości dostępnego koreceptora CCR5 na powierzchni błony komórkowej. W przypadku osób posiadających jeden zmutowany allel podatność na zakażenie i progresję choroby jest mniejsza (28, 64, 65). U osób takich obserwuje się wolniejszy spadek poziomu limfocytów CD4+, mniejszą ilość kopii wirusa w surowicy oraz lepszą odpowiedź na leczenie antyretrowirusowe (5, 66, 67). W większości publikacji opisujących wpływ wariantów genów receptorów chemokin na zakażenie HIV-1 analizowane genotypy i haplotypy zawierają allel Δ32 CCR5. Osoby będące homozygotami Δ32, są całkowicie oporne za zakażenie szczepami CCR5-tropowymymi (tzw. wirus R5), które przeważają we wczesnych etapach infekcji oraz dominują w przypadku zakażeń drogą seksualną, prawdopodobnie w wyniku większej produkcji do nasienia poprzez makrofagi i limfocyty T pamięci (T-memory cells) (17, 68, 69). Należy jednak podkreślić, że nie można uważać osób homozygotycznych Δ32/Δ32 za całkowicie pozbawione ryzyka zakażenia, gdyż są one podatne na zakażenie szczepami CXCR4-tropowymi (tzw. wirus R4). W piśmiennictwie opisywane są przypadki zakażenia takich osób z szybszą progresją zakażenia i dominacją szczepów o pojedynczym tropizmie dla receptora CXCR4 (70, 71). Prawdopodobieństwo zakażenia wirusem o tropizmie X4 jest niskie, ponieważ zarówno częstość występowania szczepów X4, jak ich zakaźność jest mniejsza, niemniej w przypadkach kiedy doszło do zakażenia, odnotowano szybszy spadek poziomu limfocytów CD4+ i kliniczną progresję zakażenia związaną z większą wirulencją wirusa in vivo (38, 72).

Ryc. 1a. Struktura receptora CCR5– typ „dziki” – niezmutowany.
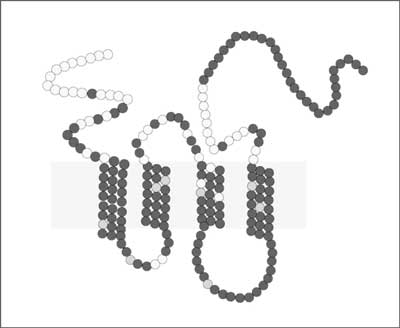
Ryc. 1b. Struktura receptora CCR5z mutacją Δ32 – utrata części domen przezbłonowych.
Rozpowszechnienie zmutowanego allelu Δ32 jest przykładem selekcji czynnika genetycznego, związanej z oddziaływaniem czynników zakaźnych na populację ludzką. Zakażenie wirusem HIV jest epidemią współczesną i nie miało wpływu na częstość występowania delecji. Istnieje hipoteza, że selekcja allelu dokonała się pod wpływem epidemii dżumy w latach 1300-1600 (73). Jednakże czas trwania epidemii był za krótki, żeby wytłumaczyć wysoką – w pewnych populacjach ponad 10% – częstość występowania allelu. Ponadto nie jest prawdopodobne, żeby utrata funkcjonalnego receptora chemokin była czynnikiem zmniejszającym śmiertelność związaną z zakażeniem Yersinia pestis. Alternatywna hipoteza sugeruje, że selekcja dokonywała się w dłuższym okresie w wyniku epidemii ospy ponad 2000 lat temu, które dotykały Europę Północną (74). Ponadto zarówno wirusy z grupy poxviridae, jak retroviridae używają receptorów chemokin do integracji z komórką docelową, co pozwala przypuszczać, że allel Δ32 mógł wywierać ochronny wpływ w przypadku zakażenia ospą.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Fowke KR, Dong T, Rowland-Jones SL et al.: HIV type 1 resistance in Kenyan sex workers is not associated with altered cellular susceptibility to HIV type 1 infection or enhanced beta-chemokine production. AIDS Res Hum Retroviruses 1998; 14: 1521-30.
2. Clegg AO, Ashtona LJ, Biti RA et al.: CCR5 promoter polymorphisms, CCR5 59029A and CCR5 59353C, are under represented in HIV-1-infected long-term non-progressors. AIDS 2000; 14: 103-108.
3. Dennis L, Kasper, Eugene Braunwald et al.: Harrison's Principles of Internal Medicine (16th Edition) 2004; 1: 1109.
4. Vidal F, Vilades C, Domingo P et al.: Spanish HIV-1-infected long-term nonprogressors of more than 15 years have an increased frequency of the CX3CR1 249I variant allele. Journal of Acquired Immune Deficiency Syndrome 2004; 37: 1534-1538.
5. Wąsik TJ, Smoleń J, Kruszyński P et al.: Associations between CCR5, CCR2 and SDF-1 genes polymorphisms and Human Immunodeficiency Virus type 1 disease progression in the Polish population. HIV & AIDS Review 2006; 5: 7-15.
6. Marmor M, Hertzmark K, Thomas SM et al.: Resistance to HIV Infection. Journal of Urban Health 2006; 83: 5-17.
7. Fauci AS: Resistance to HIV-1 infection: it's in the genes. Nat Med 1996; 2: 966-7.
8. Rowland-Jones S, Sutton JAY, Dong T et al.: HIV-specific cytotoxic T-cells in HIV-exposed but uninfected Gambian women. Nature Medicine 1995; 1: 59-64.
9. Aarons E, Fernandez M, Rees A et al.: CC-chemokine receptor 5 genotypes and in vitro susceptibility to HIV-1 of a cohort of British HIV-exposed uninfected homosexual men. AIDS 1997; 11: 688-9.
10. Alkhatib G, Combadiere C, Broder CC et al.: CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996; 272: 1955-8.
11. Berson JF, Long D, Doranz BJ et al.: A seven-transmembrane domain receptor involved in fusion and entry of T-cell-tropic Human Immunodeficiency Virus type 1 strains. Journal of Virology 1996; 70: 6288-6295.
12. Cohen J: AIDS Research: Likely HIV Cofactor Found. Science 1996; 272, 809: 809.
13. De Roda Husman AM, Koot M, Cornelissen M et al.: Association between CCR5 genotype and the clinical course of HIV-1 infection. Ann Intern Med 1997; 127: 882-90.
14. Dean M, Carrington M, Winkler C et al.: Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science 1996; 273: 1856-62.
15. Faure S, Meyer L, Costagliola D et al.: Rapid progression to AIDS in HIV+ individuals with a structural variant of the chemokine receptor CX3CR1. Science 2000; 287: 2274-7.
16. Katzenstein TL, Eugen-Olsen J, Hofmann B et al.: HIV-infected individuals with the CCR delta32/CCR5 genotype have lower HIV RNA levels and higher CD4 cell counts in the early years of the infection than do patients with the wild type. Copenhagen AIDS Cohort Study Group. J Acquir Immune Defic Syndr Hum Retrovirol 1997; 16: 10-4.
17. Liu R, Paxton WA, Choe S et al.: Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996; 86: 367-77.
18. Mariani R, Sally Wong, Mulder LCF et al.: CCR2-64I Polymorphism Is Not Associated with Altered CCR5 Expression or Coreceptor Function. Journal of Virology 1999; 73: 2450-2459.
19. An P, Nelson GW, Wang L et al.: Modulating influence on HIV/AIDS by interacting RANTES gene variants. Proceedings of National Academy of Sciences USA 2002; 99: 10002-10007.
20. Kaslow RA, Dorak T, Tang J: Influence of host genetic variation on susceptibility to HIV type 1 infection. Journal of Infectious Diseases 2005; 191 (Suppl. 1): 68-77.
21. Liu H, Chao D, Nakayama EE et al.: Polymorphism in RANTES chemokine promoter affects HIV-1 disease progression. Proceedings of National Academy of Sciences USA 1999; 96: 4581-4585.
22. Magierowska M, Theodorou I, Debre P et al.: Combined genotypes of CCR5, CCR2, SDF1, and HLA genes can predict the long-term nonprogressor status in Human Immunodeficiency Virus-1 – infected individuals. Blood 1999; 93: 936-941.
23. Nakayama EE, Hoshino Y, Xin X et al.: Polymorphism in the interleukin-4 promoter affects acquisition of Human Immunodeficiency Virus type 1 syncytium-inducing phenotype. Journal of Virology 2000; 74: 5452-5459.
24. Shin HD, Winkler C, Stephens JC et al.: Genetic restriction of HIV-1 pathogenesis to AIDS by promoter alleles of IL10. Proceedings of National Academy of Sciences USA 2000; 97: 14467-14472.
25. Vasilescu A, Heath SC, Ivanova R et al.: Genomic analysis of Th1-Th2 cytokine genes in an AIDS cohort: identification of IL4 and IL10 haplotypes associated with the disease progression. Genes and Immunity 2003; 4: 441-444.
26. Winkler C, Modi W, Smith MW et al.: Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. Science 1998; 279: 389-393.
27. Puissant B, Roubinet F, Massip P et al.: Analysis of CCR5, CCR2, CX3CR1, and SDF1 polymorphisms in HIV-positive treated patients: impact on response to HAART and on peripheral T lymphocyte counts. AIDS Research and Human Retroviruses 2006; 22: 153-162.
28. Huang Y, Paxton WA, Wolinsky SM et al.: The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med 1996; 2: 1240-3.
29. Samson M, Libert F, Doranz BJ et al.: Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996; 382: 722-725.
30. Rowe PM: CKR-5 deletion heterozygotes progress slower to AIDS. Lancet 1996; 348: 947.
31. Quillent C, Oberlin E, Braun J et al.: HIV-1-resistance phenotype conferred by combination of two separate inherited mutations of CCR5 gene. Lancet 1998; 351: 14-8.
32. Shrestha S, Strathdee SA, Galai N et al.: Behavioral risk exposure and host genetics of susceptibility to HIV-1 infection. Journal of Infectious Diseases 2006; 193: 16-26.
33. Hladik F, Liu H, Speelmon E et al.: Combined effect of CCR5-Δ32 heterozygosity and the CCR5 promoter polymorphism -2459 A/G on CCR5 expression and resistance to Human Immunodeficiency Virus type 1 transmission. Journal of Virology 2005; 79: 11677-11684.
34. McDermott DH, Zimmerman PA, Guignard F et al.: CCR5 promoter polymorphism and HIV-1 disease progression. Lancet. 1998; 352: 866-70.
35. Kostrikis LG, Avidan U, Neumann et al.: A polymorphism in the regulatory region of the CC-Chemokine receptor 5 gene influences perinatal transmission of Human Immunodeficiency Virus Type 1 to African-American infants. Journal of Virology 1999; 73: 10264-10271.
36. Nakayama EE, Tanaka Y, Nagai Y et al.: A CCR2-V64I polymorphism affects stability of CCR2A isoform. AIDS 2004; 18: 729-738.
37. Tang J, Shelton B, Makhatadze NJ et al.: Distribution of chemokine receptor CCR2 and CCR5 genotypes and their relative contribution to human immunodeficiency virus type 1 (HIV-1) seroconversion, early HIV-1 RNA concentration in plasma, and later disease progression. J Virol 2002; 76: 662-72.
38. Michael NL, Louie LG, Rohrbaugh AL et al.: The role of CCR5 and CCR2 polymorphisms in HIV-1 transmission and disease progression. Nat Med 1997; 3: 1160-2.
39. Smith MW, Dean M, Carrington M et al.: Contrasting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC), ALIVE Study. Science 1997; 277: 959-65.
40. Kostrikis LG, Huang Y, Moore JP et al.: A chemokine receptor CCR2 allele delays HIV-1 disease progression and is associated with a CCR5 promoter mutation. Nat Med 1998; 4: 350-3.
41. Karam W, Jurjus R, Khoury N et al.: Frequency of the CCR5-delta 32 chemokine receptor gene mutation in the Lebanese population. East Mediterr Health J 2004; 10: 671-5.
42. Vidal F, Vilades C, Domingo P et al.: Chemokines LTNP Study Group. Spanish HIV-1-infected long-term nonprogressors of more than 15 years have an increased frequency of the CX3CR1 249I variant allele. J Acquir Immune Defic Syndr 2005; 40: 527-31.
43. Trachtenberg E, Korber B, Sollars C et al.: Advantage of rare HLA supertype in HIV disease progression. Nat Med 2003; 9: 928-35.
44. Kulkarni S, Tripathy S, Agnihotri K et al.: Indian primary HIV-2 isolates and relationship between V3 genotype, biological phenotype and coreceptor usage. Virology 2005; 337: 68-75.
45. Anastassopoulou CG, Kostrikis LG: The impact of human allelic variation on HIV-1 disease. Current HIV Research 2003: 185-203.
46. MacDonald KS, Fowke KR, Kimani J et al.: Influence of HLA supertypes on susceptibility and resistance to Human Immunodeficiency Virus type 1 infection. Journal of Infectious Diseases 2000; 181: 1581-9.
47. Sriwanthana B, Hodge T, Mastro TD et al.: HIV-specific cytotoxic T lymphocytes, HLA-A11, and chemokine-related factors may act synergistically to determine HIV resistance in CCR5 delta32-negative female sex workers in Chiang Rai, northern Thailand. AIDS Res Hum Retroviruses 2001; 17: 719-34.
47. Winchester R, Chen Y, Rose S, Selby JWB: Major histocompatibility complex class II DR alleles DRB1*1501 and those encoding DR13 are preferentially associated with a diminution in maternally transmitted HIV-1 infection in different ethnic groups: determination by an automated sequence-based typing method. Proceedings of National Academy of Sciences USA 1995; 92: 12374-8.
48. Borghans JA, M?lgaard A, de Boer RJ, Keşmir C: HLA alleles associated with slow progression to AIDS truly prefer to present HIV-1 p24. PLoS ONE 2007; 19: e920.
49. Singh KK, Hughes MD, Chen J, Spector SA: Lack of protective effects of interleukin-4-589-C/T polymorphism against HIV-1-related disease progression and central nervous system impairment, in children. Journal of Infectious Diseases 2004; 189: 587-592.
50. Baggiolini M: Chemokines and leukocyte traffic. Nature 1998; 392: 565-568.
51. Le Y, Zhou Y, Iribarren P, Wang JM: Chemokines and Chemokine Receptors: Their Manifold Roles in Homeostasis and Disease. Cellular & Molecular Immunology 2004; 1: 95-104.
52. Negus RPM: The chemokines: cytokines that direct leukocyte migration. Journal of the Royal Society of Medicine 1996; 89: 312-314.
53. Murphy PM: International Union of Pharmacology. Update on chemokine receptor nomenclature. Pharmacological Reviews 2002; 54: 227-229.
54. Rollins BJ: Chemokines. Blood 1997; 90: 909-928.
55. Hogan CM, Hammer SM: Host determinans in HIV infection and disease part two Ann Intern Med 2001; 134: 978-976.
56. Hogan CM, Hammer SM: Host determinans in HIV infection and disease. part one Ann Intern Med 2001; 134: 761-76.
57. D'Souza MP, Cairns JS, SF P: Current evidence and future directions for targeting HIV entry: therapeutic and prophylactic strategies. JAMA 2000; 2: 215-222.
58. Kawamura T, Bruse SE, Abraha A et al.: PSC-RANTES blocks R5 human immunodeficiency virus infection of Langerhans cells isolated from individuals with a variety of CCR5 diplotypes. J Virol 2004; 78: 7602-9.
59. McDermott D, Colla JS, Kleeberger CA et al.: Genetic polymorphism in CX3CR1 and risk of HIV disease. Science 2000; 290: 2031.
60. Winkler CA, Hendel H, Carrington M et al.: Dominant effects of CCR2–CCR5 haplotypes in HIV-1 disease progression. Journal of Acquired Immune Deficiency Syndrome 2004; 37: 1534-1538.
61. Capoulade-Metay C, Ma L, Truong LX et al.: New CCR5 variants associated with reduced HIV coreceptor function in southeast Asia. AIDS 2004; 18: 2243-2252.
62. Eugen-Olsen J, Iversen AK, Benfield TL et al.: Chemokine receptor CCR2b 64I polymorphism and its relation to CD4 T-cell counts and disease progression in a Danish cohort of HIV-infected individuals. Copenhagen AIDS cohort. J Acquir Immune Defic Syndr Hum Retrovirol 1998; 18: 110-6.
63. Glass WG, McDermott DH, Lim JK et al.: CCR5 deficiency increases risk of symptomatic West Nile virus infection. Journal of Experimental Medicine. 2005. Dostęp on-line www.jem.org/cgi/doi/10.1084/jem.20051970).
64. Rappaport J, Cho YY, Hendel H et al.: 32 bp CCR-5 gene deletion and resistance to fast progression in HIV-1 infected heterozygotes. Lancet 1997; 349: 922-3.
65. Parczewski M, Leszczyszyn-Pynka M, Kaczmarczyk M et al.: Sequence variants of chemokine receptor genes and susceptibility to HIV-1 infection. J Appl Genet 2009; 50: 159-166.
66. Morawetz RA, Rizzardi GP, Glauser D et al.: Genetic polymorphism of CCR5 gene and HIV disease: the heterozygous (CCR5/delta ccr5) genotype is neither essential nor sufficient for protection against disease progression. Swiss HIV Cohort. Eur J Immunol 1997; 27: 3223-7.
67. Tang J, Kaslow RA: The impact of host genetics on HIV infection and disease progression in the era of highly active antiretroviral therapy. AIDS 2003; 17: 51-60.
68. Smith MW, Carrington M, Winkler C et al.: CCR2 chemokine receptor and AIDS progression. Nat Med 1997; 3: 1052-3.
69. Stewart GJ, Ashton LJ, Biti RA et al.: Increased frequency of CCR-5 delta 32 heterozygotes among long-term non-progressors with HIV-1 infection. The Australian Long-Term Non-Progressor Study Group. AIDS 1997; 11: 1833-8.
70. O'Brien TR, Winkler C, Dean M et al.: 2nd. HIV-1 infection in a man homozygous for CCR5 delta 32. Lancet 1997; 349: 1219.
71. Theodorou I, Meyer L, Magierowska M et al.: HIV-1 infection in an individual homozygous for CCR5 delta 32. Seroco Study Group. Lancet 1997; 349: 1219-20.
72. Michael NL, Nelson JAE, Kewalramani VN et al.: Exclusive and persistent use of the entry coreceptor CXCR4 by Human Immunodeficiency Virus type 1 from a subject homozygous for CCR5 Δ32. Journal of Virology 1998; 72: 6040-6047.
73. Galvani AP, Novembre J: The evolutionary history of the CCR5-Δ32 HIV-resistance mutation. Microbes and Infection 2005; 7: 302-309.
74. Galvani AP, Slatkin M: Evaluating plague and smallpox as historical selective pressures for the CCR5-Δ32 HIV-resistance allele. Proceedings of National Academy of Sciences USA 2003; 100: 15276-15279.
75. Martin MP, Dean M, Smith MW et al.: Genetic acceleration of AIDS progression by a promoter variant of CCR5. Science 1998; 282: 1907-11.
76. Mummidi S, Bamshad M, Ahuja SS et al.: Evolution of Human and non-human Primate CC chemokine receptor 5 gene and mRNA. Journal of Biological Chemistry 2000; 275: 18946-18961.
77. Mangano A, Gonzalez E, Dhanda R et al.: Concordance between the CC chemokine receptor 5 genetic determinants that alter risks of transmission and disease progression in children exposed perinatally to Human Immunodeficiency Virus. The Journal of Infectious Diseases 2001; 183: 1574-1585.
78. Mulherin SA, O'Brien TR, Ioannidis JPA et al.: Effects of CCR5-Δ32 and CCR2-64I alleles on HIV-1 disease progression: the protection varies with duration of infection. AIDS 2003; 17: 377-387.
79. Meyer L, Magierowska M, Hubert JB et al.: Early protective effect of CCR-5 delta 32 heterozygosity on HIV-1 disease progression: relationship with viral load. The SEROCO Study Group. AIDS 1997; 11: 73-8.
80. Lee B, Doranz BJ, Rana S et al.: Influence of the CCR2-V64I Polymorphism on Human Immunodeficiency Virus Type 1 Coreceptor Activity and on Chemokine Receptor Function of CCR2b, CCR3, CCR5, and CXCR4. Journal of Virology 1998; 72: 7450-7458.
81. Umehara H, Bloom ET, Nagano TOY et al.: Fractalkine in vascular biology – from basic research to clinical disease. Arterioscler Thromb Vasc Biol 2004; 24: 34-40.
82. Combadie"re C, Potteaux S, Gao J-L et al.: Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation 2003; 107: 1009-1016.
83. Moatti D, Faure S, Fumeron F et al.: Polymorphism in the fractalkine receptor CX3CR1 as a genetic risk factor for coronary artery disease. Blood 2001; 97: 1925-1928.
84. Foussat A, Bouchet-Delbos L, Berrebi D et al.: Deregulation of the expression of the fractalkine/fractalkine receptor complex in HIV-1-infected patients. Blood 2001; 98: 1678-1686.
85. Becker Y: The spreading of HIV-1 infection in the human organism is caused by fractalkine trafficking of the infected lymphocytes-a review, hypothesis and implications for treatment. Virus Genes 2006. publikacja on-line.
