© Borgis - New Medicine 1/2013, s. 14-20
*Paweł Kowalczyk1, Jolanta Krzyczkowska2, Urszula Jankiewicz3
Mechanisms of the dna repair in bacterial and yeast cells
1Autonomous Department of Microbial Biology, Faculty of Agriculture and Biology, Warsaw University of Life Sciences, Warsaw, Poland
2Department of Chemistry, Faculty of Food Sciences, Warsaw University of Life Sciences, Warsaw, Poland
3Department of Biochemistry, Warsaw University of Life Sciences, Warsaw, Poland
Summary
Malfunctioning of some DNA repair pathways predisposes to certain types of cancer. Impaired base excision repair, nucleotide excision repair, mismatch repair and recombination are implied in human tissues. These repair pathways are engaged very quickly in the cell when the external (chemical carcinogens) and internal (lipid peroxidation products) compounds react directly with DNA. This reaction may either lead to different modifications (damages) in DNA and genes. This damages may lead finally to mutation and cancer progression and induce different DNA repair mechanisms such as. BER, NER, HR and MMR. In the BER mechanisms repair is initiated by the action of a damage-specific DNA N-glycosylase that is responsible for the recognition and removal of an altered base through cleavage of the N-glycosylic bond and action of AP-endonuclease. Nucleotide excision repair (NER) is the most versatile and flexible DNA repair pathway of living cells as it deals with a wide range of structurally unrelated DNA lesions. NER corrects a wide array of DNA lesions that distort the DNA double helix, interfere in base pairing and block DNA duplication and transcription. The most common examples of these lesions are the cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4 PPs) induced by ultraviolet radiation (UV) and bases with large substitutes derived from chemicals such as polycyclic aromatic hydrocarbons or exocyclic adducts. Homologous recombination, utilize large regions of DNA homology, usually the homological chromosome, to exchange damaged DNA for the intact one. DNA mismatch-repair system (MMR) is involved in the repair of mispaired bases formed during replication, genetic recombination and as a result of DNA damage.
INTRODUCTION
Different mechanisms of the DNA repair
To counteract deleterious consequences of DNA damage, the cells developed several repair mechanisms which eliminate from genomes mis-instructive or non-instructive elements, as well as seal DNA breaks. Various repair systems can be classified into the following groups, according to repair mechanism:
1. Sanitisation of nucleotide pool from modified nucleoside triphosphates, which prevents their incorporation into DNA by DNA polymerases.
2. Direct reversal of DNA damage, which occurs without breaking of the double helix.
3. Excision repair, in which a fragment of DNA strand containing the lesion is excised, followed by DNA resynthesis on the template of the opposite intact strand. Three different systems utilize the excision mode for DNA repair:
– Mismatch repair, which eliminates replication errors, some damaged DNA bases, as well as small loops.
– Base excision repair, initiated by DNA glycosylases which cleave out the damaged base and initiate the synthesis step.
– Nucleotide excision repair, in which a larger fragment of damaged DNA strand is removed (12-13 nucleotides in E. coli, 24-32 in eucaryota).
4. Recombination, which is working in the situation of gross damage and unavailability of intact opposite DNA strand or for double-strand break (DSB) repair.
5. SOS repair, based on translesion synthesis past DNA damages by specified class of low fidelity DNA polymerases tolerant of template structure abnormality, but introducing errors into DNA. Repair of exocyclic, unsubstituted etheno-DNA adducts is realized mainly by base excision repair pathway (1). 1,N2-propanoguanosine with a hexyl side chain derived from hydroxynonenal interaction with DNA was shown to be eliminated by nucleotide excision repair in mammalian cells (2, 3).
Base excision repair
Base excision repair constitutes the primary defense against lesions that do not heavily distort the DNA structure. BER is responsible for the removal of a variety of lesions. These include spontaneous hydrolytic depurination of DNA, deamination of bases, products of reaction with hydroxyl radicals, and covalent DNA adducts formed by intracellular LPO and small reactive metabolites, such as methylating agents. Repair is initiated by the action of a damage-specific DNA N-glycosylase that is responsible for the recognition and removal of an altered base through cleavage of the N-glycosylic bond (4, 5). Base removal generates an apurinic/apyrimidinic (AP) site, a noncoding DNA lesion that is both cytotoxic and mutagenic. The abasic site is the substrate for an AP endonuclease that hydrolyzes the phosphodiester bond 5” to an abasic site (6-9). Cleavage by AP-endonuclease generates a 3”-OH terminus suitable for extension by a DNA polymerase. The resulting 5” terminus contains a deoxyribose phosphate residue (dRP), which must be removed and replaced in order to complete repair. BER can proceed via two pathways, designated short patch (10) or long patch repair (11, 12). In the short patch repair pathway DNA polymerase β adds a single nucleotide and also cleaves the 5”-deoxyribose phosphate residue using an intrinsic deoxyribosephosphate lyase (dRP lyase) activity (13, 14). A DNA ligase then seals the nick to complete repair. This generates a repair product where only the damaged nucleotide is replaced. In the long patch DNA polymerase δ/ε or β and PCNA extend DNA chain at the 3”-OH terminus generated by an AP-endonuclease, displacing the strand at the 5” end. The antibodies directed against PCNA totally suppress repair patches longer than one nucleotide. This creates a baseless sugar-containing flap, removed by a human flap-endonuclease (FEN1) which is also engaged in excision of primers during replication. DNA ligase I in long patch or ligase III in the short patch seal phosphodiester bonds.
DNA N-glycosylases
DNA glycosylases are often able to act upon a variety of DNA adducts that result from the action of a number of DNA damaging agents, although with different efficiencies and sometimes overlapping specificities. The lesions recognized by DNA glycosylases include noncanonical Watson-Crick base pairs and bases altered by deamination, oxidation and alkylation. On the basis of their primary substrate specificity all identified DNA glycosylases have been classified into several types, namely, DNA glycosylases of deaminated, mismatched, alkylated, and oxidized bases as well as of pyrimidine-dimers. DNA glycosylases are generally globular, monomeric, small proteins, ranging in molecular mass from 16 to 60 kDa. They often contain a conserved motif of helix-harpin-helix (HhH) which enables them to bind DNA. DNA glycosylases act on damaged bases by flipping damaged nucleotides out of the DNA into an enzyme active site pocket, where the excision takes place (15-17). DNA glycosylases can be separated into two groups: 1) enzymes that possess only N-glycosylase activity to generate an AP site (monofunctional DNA glycosylases), and 2) proteins that possess DNA glycosylase activity and an activity to incise the phosphodiester backbone immediately 3” of the resulting AP site via b-elimination, or 3” and 5” via b-δ-elimination, resulting in a single nucleotide gap flanked by phosphate termini (3”/5”) (bifunctional glycosylases/AP lyases) (18). Due to an associated lyase activity, bifunctional glycosylases are sometimes termed endonucleases. Cloning of the OGG1 gene from Saccharomyces cerevisiae has revealed that DNA glycosylases are not necessarily conserved throughout phylogeny, yet there is a DNA-repair protein superfamily with a wide substrate specificity found from bacteria to man.
Nucleotide excision repair (NER)
Nucleotide excision repair (NER) is the most versatile and flexible DNA repair pathway of living cells as it deals with a wide range of structurally unrelated DNA lesions. NER corrects a wide array of DNA lesions that distort the DNA double helix, interfere in base pairing and block DNA duplication and transcription. The most common examples of these lesions are the cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4 PPs), induced by ultraviolet radiation (UV), and bases with large substitutes derived from chemicals such as polycyclic aromatic hydrocarbons (19). However, NER can also correct smaller modified bases. This is the major pathway responsible for removing from DNA such exocyclic adducts as M1G and 1,N2-propanoguanine (20, 21). The overall processes of NER in eukaryotic and prokaryotic cells are similar, but there are many differences in detail. NER can be separated into two subpathways, slow, global genome repair (GGR) and fast, transcription-coupled repair (TCR). Global repair is the process by which most lesions are repaired regardless of their location in the genome. Transcription-coupled repair is characterized by the more rapid repair of lesions in the transcribed strand of an expressed gene than in the nontranscribed strand or in the rest of the genome (22). The mechanism of these two pathways is mainly similar except for recognition of the damage and, therefore, for initiation of the process. In E. coli, both subpathways require the full set of NER proteins, but transcription-coupled repair additionally requires an actively transcribing RNA polymerase (RNAP) and at least one additional factor, the transcription repair coupling factor, encoded by the mfd gene (23). The latter factor is thought to recruit Uvr proteins to RNAP arrested at a lesion on the transcribed strand (24), resulting in rapid repair of the transcription-blocking lesion.
NER in E. coli requires six proteins: UvrA, UvrB, UvrC, UvrD, DNA polymerase I, and ligase (25). In vivo, UvrA is present both as a monomer and a dimer, the latter complexing with UvrB for initial DNA damage recognition. This UvrA2B heterotrimer may carry out limited, ATP-dependent, processive scanning of the damaged region until the actual damage site is encountered (26). At this point, a conformational change occurs in the protein-DNA complex, leading to release of the UvrA dimer, stable UvrB-DNA binding, and a local bending and unwinding of the damaged region of DNA. UvrC then binds to the UvrB-DNA complex, unmasking the cryptic endonuclease activity of UvrB. In the case of UV photoproducts, this activity causes an incision to be made four bases 3” from the lesion. A second incision is made by the UvrBC complex seven bases 5” from the lesion. UvrD, also known as DNA helicase II, releases UvrC and the oligonucleotide between the dual incisions, leaving UvrB at a 12-base gap on one strand. DNA polymerase I fills the gap and dissociates the UvrB protein from the DNA. The repair process is completed by DNA ligase, which seals the nick.
In eukaryotes, most of NER studies were performed with cells mutated at different steps of the pathway, including cells from human patients with genetic syndromes directly related to DNA repair, such as Cockayne”s syndrome (CS), trichothiodystrophy (TTD) and xeroderma pigmentosum (XP). Xeroderma pigmentosum is marked by an increased sensitivity to light, leading to an increase in skin carcinomas, and a number of neurological abnormalities (27). There are seven complementation groups for xeroderma pigmentosum, XPA to XPG, defined by the ability of fused fibroblasts to repair damage after UV irradiation. The genes mutated in these complementation groups have been identified, and are homologues of yeast NER genes (28) Cockayne”s syndrome and trichothiodystrophy have overlapping symptoms with xeroderma pigmentosum, such as neurological degeneration, and some of the XP complementation groups overlap with the CS and TTD groups.
In vitro reconstitution of the NER process has defined six core factors, some with multiple subunits, that are required for the repair of most damage in DNA (29). These proteins are XPA, the RPA hetero-trimer, the XPC-hHR23B complex, the 6-9 subunit TFIIH complex and the two structure-specific endonucleases, XPG and the heterodimeric ERCC1-XPF. Human NER involves the formation of an open, unwound intermediate, which spans 24-32 bp around the lesion. The formation of this open complex is ATP dependent, and the earliest stages of opening absolutely require XPC-hHR23B and TFIIH, which contains two helicases of opposing polarities, XPB and XPD. Full opening to give a structure that can be incised by XPG and ERCC1-XPF requires the presence of most of the core NER factors. XPA, RPA, XPC-hHR23B, TFIIH and XPG are involved, although the catalytic activity of XPG is not a requirement of this stage (30). Incision of the damaged strand on the 3” side of a lesion is made by XPG and usually occurs before 5” incision by ERCC1-XPF (31).Dual incision results in the release of a damage-containing oligonucleotide 24-32 residues in length (32). The gap is then filled by DNA polymerase δ or ε holoenzyme (33) and sealed by a DNA ligase (fig. 1).
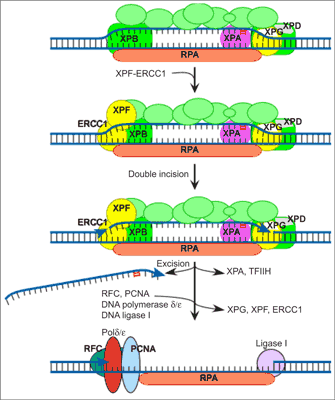
Fig. 1. Model of nucleotide excision repair.
Recombination
Both in bacteria and mammalian cells two different modes of recombination can be distinguished: 1) homologous recombination, which utilize large regions of DNA homology, usually the homological chromosome, to exchange damaged DNA for the intact one, 2) non-homologous recombination, in which double-strand breaks (DSBs) of different chromatin regions are joined together on the basis of microhomologies to produce a new gene configuration. In mammals this system is also utilized for the production of antibodies. In bacteria the main repair mode is homologous recombination, while in mammals both systems are used by the cell.
Homologous recombination in E. coli
At least 25 different proteins are involved in all types of homologous recombination in E. coli (34); these include the RecA, RecBCD, RecF, RecG, RecJ, RecN, RecO, RecQ, RecR, RuvAB, RuvC, PriA and SSB proteins, DNA polymerases, DNA topoisomerases and DNA ligase, as well as the cis-acting recombination hotspot χ. Many of these proteins have functional homologs in other bacteria, Eucaryota, Archaea and some phages. In fact, a RecA-like protein is present in all free-living organisms (35). The yeast Saccharomyces cerevisiae is an ideal model organism for studying these repair processes.
Purified RecA protein can homologously pair and exchange DNA strands in vitro. One of the most useful model systems, the three-strand DNA exchange reaction, utilizes circular single-stranded DNA (ssDNA) and homologous linear duplex DNA substrates. In the presence of a nucleotide cofactor RecA protein polymerizes on the ssDNA to form a helical nucleoprotein filament (36). This nucleoprotein filament both aligns and pairs with a homologous region in the duplex DNA to form joint molecules. Consequently, the RecA protein filament must accommodate two DNA molecules and must bring them sufficiently close together to promote exchange of DNA strands (36). DSBs induced directly by ionizing radiation, and indirectly as a natural consequence of DNA replication on a chemically flawed template, are lethal and need repair via recombination pathway. The recombinational repair process consists of four steps: 1) initiation, 2) homologous pairing and DNA strand exchange, 3) DNA heteroduplex extension (branch migration), and 4) resolution (fig. 2).
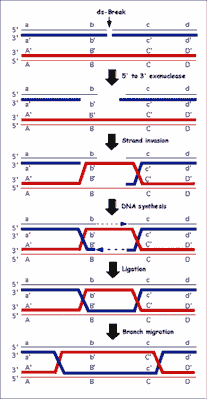
Fig. 2. General model for homologous recombination in all organisms. The model depicted is the double-strand-break repair (DSBR) model. Dark gray lines indicate newly synthesized DNA.
The first step, initiation, represents the linear duplex DNA at the DSB to produce the ssDNA needed for DNA strand invasion of a dsDNA homolog by RecA protein. For the prototypic homologous pairing reactions promoted by RecA protein, ssDNA is a prerequisite (37). For the second step of recombination, DNA strand exchange, to occur between two homologous dsDNA molecules, processing of one duplex to produce a region of ssDNA is conventionally invoked. This processing involves the recombination-specific helicases, the RecBCD and RecQ proteins, with the latter thought to work in conjunction with the RecJ exonuclease. RecBCD enzyme is a DNA helicase that also possesses a 3”→5” nuclease activity on the strand with the χ sequence, as well as stimulation of the 5”- to 3”-nuclease activity responsible for degrading the opposite strand of the duplex (38, 39). RecBCD is regulated by the direct interaction with the recombination hotspot χ. Recombination hotspots in E. coli, known as Chi sites (χ = 5”-GCTGGTGG-3”) enhance the frequency of recombination in their vicinity 5-10-fold (40). Modification of RecBCD enzymatic activity (ssDNA exonuclease, ssDNA endonuclease, dsDNA exonuclease, DNA dependent ATP-ase and DNA helicase) by χ is coordinated with the loading of RecA protein onto the χ-containing ssDNA by RecBCD enzyme, ensuring incorporation of this ssDNA into a recombinationally proficient nucleoprotein complex. RecQ protein is also a DNA helicase; in wild-type cells it functions in the so-called RecF pathway, which can also act efficiently at DSBs when the RecBCD is rendered non-functional by mutation (41). If RecA protein fails to assemble on the ssDNA produced, then accessory proteins RecF, RecO and RecR facilitate this assembly step (42). Upon assembly of a contiguous RecA protein filament on ssDNA, called the presynaptic filament, subsequent homology search can ensue. The third step of recombination is DNA heteroduplex extension; here, a specialized motor protein complex, the RuvAB complex, functions. The RuvAB complex is a DNA helicase that extends the region of DNA heteroduplex by branch migrating the crossover point (43). The final step of recombination requires separation of the two DNA molecules. This important resolution step is left to a Holliday junction-specific endonuclease, the RuvC protein. The RuvC protein, as part of a complex with the RuvAB proteins, recognizes and cleaves Holliday junctions to complete the recombination process.
DNA Mismatch repair (MMR) pathway
DNA mismatch-repair system is involved in the repair of mispaired bases formed during replication, genetic recombination and as a result of DNA damage. DNA lesions may be recognized and repaired by more than one DNA-repair process. If two repair systems with different error frequencies have overlapping lesion specificity and one or both is inducible, the resulting variable competition for the lesions can change the biological consequences of these lesions. This concept was demonstrated by observing mutation in yeast cells (Saccharomyces cerevisiae) exposed to combinations of mutagens under conditions which influenced the induction of error-free recombinational repair or error-prone repair. Studies in yeast indicate the involvement of the mismatch repair pathway in prevention of genotoxic effect of oxidative DNA damage. Bacterial cells use differential methylation of the two strands in order to differentiate between “right” or “wrong” undamaged normal base in each strand. E. coli MMR system consists of three genes: mutH, mutL, mut S (44). The first step is the detection of a mismatch through its binding by MutS (45). MutS recruits MutL and together they activate MutH. The communication between MutS and MutH was mediated by MutL in an ATP hydrolysis-dependent manner (46). No biological activity for the MutL protein has been identified, but it may act as “molecular matchmaker”, coupling mismatch recognition by MutS to MutH. MutH cleaves the newly replicated error-containing daugther strand at a transiently unmethylated d(GATC) site. The degraded strand is replaced by DNA polymerase III holoenzyme which is assisted by single-strand binding proteins, with repair being completed by ligation of the resulting nick by DNA ligase I. Mammalian MMR consists of seven proteins, including three bacterial MutS homologs: hMSH2, hMSH3 and hMSH6, and four MutL homologs, hMLH1, hMLH3, hPMS1, and hPMS2. MMR proteins function as heterodimeric complexes. For example, the hMSH2-hMSH6 heterodimer recognizes single base pair mismatches and small insertion/deletion loops. For example the complex binds to 8oxoG:A mispairs and 8oxoG:C base pairs with high affinity and specificity. Mutations in hMSH2 and hMSH6 caused a synergistic increase in mutation rate in combination with mutations in OGG1, increase 218-fold in GC→TA transversion, (47). The heterodimer hMSH2-hMSH3 recognizes larger loops, containing up to 12 nucleotides (48). hMutS plays a central role in initiating mispair recognition and binding. hMutL acts as a “molecular matchmaker” between hMutS and downstream proteins (helicase and exonuclease) to complete the repair process. Excision of nucleotides proceeds from the nick to around 100 nucleotides past the mismatch. DNA polymerase α, ε or δ and DNA ligase fill the gap and seal the nick (49). Post replicative mismatch repair increases the fidelity of DNA replication from 100 to 1000-fold in E. coli. MutS-dependent MMR was also shown to correct M1G and PdG containing DNA (50) thus providing cells with a protective strategy before oxidative stress (fig. 3).
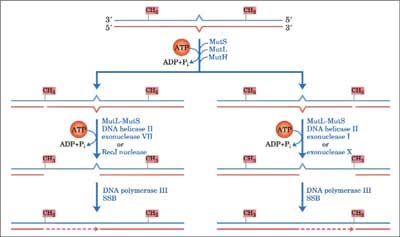
Fig. 3. General model for DNA mismatch-repair system.
These all mechanisms of DNA repair base excision repair, nucleotide excision repair, mismatch repair and recombination are implied also in human tissues. However their mutual interactions in human cells are fairly unknown (51, 52).
Piśmiennictwo
1. Gros L, Ishchenko AA, Saparbaev M: Enzymology of repair of etheno-adducts. Mutat Res 2003; 531(1-2): 219-229. 2. Chung FL, Zhang L, Ocando JE et al.: Role of 1,N2-propanodeoxyguanosine adducts as endogenous DNA lesions in rodens and humans, IARC Scientific Publication 1999; 150, IARC, Lyon, 45-54. 3. Chung FL, Nath RG, Ocando J et al.: Deoxyguanosine adducts of t-4-hydroxy-2-nonenal are endogenous DNA lesions in rodents and humans: detection and potential sources. Cancer Res 2000; 60: 1507-1511. 4. Krokan HE, Standal R, Slupphaug G: DNA glycosylases in the base excision repair of DNA. Biochem J 1997; 325: 1-16. 5. Krokan HE, Nilsen H, Skorpen F et al.: Base excision repair of DNA in mammalian cells. FEBS Lett 2000; 476(1-2): 73-77. 6. Kane CM, Linn S: Purification and characterization of an apurinic/apyrimidinic endonuclease from HeLa cells. J Biol Chem 1981; 256: 3405-3414. 7. Demple B, Herman T, Chen DS: Cloning and expression of APE, the cDNA encoding the major human apurinic endonuclease: definition of a family of DNA repair enzymes. Proc Natl Acad Sci USA 1991; 88: 11450-11454. 8. Robson CN, Hickson ID: Isolation of cDNA clones encoding a human apurinic/ /apyrimidinic endonuclease that corrects DNA repair and mutagenesis defects in E. coli xth (exonuclease III) mutants. Nucleic Acids Res 1991; 19(20): 5519-5523. 9. Seki S, Hatsushika M, Watanabe S et al.: cDNA cloning, sequencing, expression and possible domain structure of human APEX nuclease homologous to Escherichia coli exonuclease III. Biochim. Biophys Acta 1992; 1131: 287-299. 10. Kubota Y, Nash RA, Klungland A et al.: Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase β and the XRCC1 protein. EMBO J 1996; 15: 6662-6670. 11. Fortini P, Pascucci B, Parlanti E et al.: Different DNA polymerases are involved in the short- and long-patch base excision repair in mammalian cells. Biochemistry 1998; 37(11): 3575-3580. 12. Frossina G, Fortini P, Rossi O et al.: Two pathways for base excision repair in mammalian cells. J Biol Chem 1996; 271: 9573-9578. 13. Bennett RA, Wilson DM, Wong D et al.: Interaction of human apurinic endonuclease and DNA polymerase in the base excision repair pathway. Proc Natl Acad Sci USA 1997; 94: 7166-7169. 14. Podlutsky AJ, Dianova II, Wilson SH et al.: DNA synthesis and dRPase activities of polymerase are both essential for single-nucleotide patch base excision repair in mammalian cell extracts. Biochemistry 2001; 40: 809-813. 15. Lau AY, Scharer, Samson L et al.: Crystal structure of a human alkylbase- DNA repair enzyme complexed to DNA: Mechanisms for Nucleotide flipping and base excision. Cell 1998; 95: 249-258. 16. Stivers JT, Pankiewicz KW, Watanabe KA: Kinetic mechanism of damage site recognition and uracil flipping by Escherichia coli uracil DNA glycosylase. Biochemistry 1999; 38: 952-963. 17. Hollis T, Ichikawa Y, Ellenberger T: DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J 2000; 19: 758-766. 18. Dodson ML, Michaels ML, Lloyd RS: Unified catalytic mechanism for DNA glycosylases. J Biol Chem 1994; 269: 32709-32712. 19. de Laat WL, Jaspers NG, Hoeijmakers JH: Molecular mechanism of nucleotide excision repair. Genes Dev 1999; 13: 768-785. 20. Fink SP, Reddy GR, Marnett LJ: Mutagenicity in Escherichia coli of the major DNA adduct derived from the endogenous mutagen malondialdehyde. Proc Natl Acad Sci USA 1997; 94: 8652-8657. 21. Johnson KA, Fink SP, Marnett LJ: Repair of propanodeoxyguanosine by nucleotide excision repair in vivo and in vitro. J Biol Chem 1997; 272: 11434-11438. 22. Mellon I, Hanawald PC: Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature 1989; 342: 95-98. 23. Selby CP, Witkin EM, Sancar A: Escherichia coli mdf mutant deficient in “mutation frequency decline” lacks strand-specific repair: in vivo complementation with purified coupling factor. Proc Natl Acad Sci. USA 1991; 88: 11574-11578. 24. Selby CP, Sancar A: Molecular mechanism of transcription repair coupling. Science 1993; 260: 53-58. 25. Friedberg EC, Walker GC, Siede W: DNA Repair and Mutagenesis. American Society of Microbiology Press 1995; Washington, DC. 26. Grossman L, Thiagalingam S: Nucleotide excision repair, a tracking mechanism in search of damage. J Mol Biol 1993; 268: 16871-16874. 27. Lehmann AR: DNA repair-deficient diseases, Xeroderma pigmentosum, Cockayne”s syndrome and trichothiodistrophy. Biochimie 2003; 85(11); 1101-1111. 28. Aboussekhra A, Biggerstaff M, Shivji MK et al.: Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 1995; 80(6): 859-868. 29. Mu D, Hsu DS, Sancar A: Reaction mechanism of human DNA repair excision nuclease. J Biol Chem 1996; 271(14): 8285-8294. 30. Constantinou A, Gunz D, Evans E et al.: Conserved residues of human XPG protein important for nuclease activity and function in nucleotide excision repair. J Biol Chem 1999; 274(9): 5637-5648. 31. O”Donovan A, Davies AA, Moggs JG et al.: XPG endonuclease makes the 3” incision in human DNA nucleotide excision repair. Nature 1994; 371(6496): 432-435. 32. Moggs JG, Yarema KJ, Essigmann JM et al.: Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3-intrastrand d(GpTpG)-cisplatin adduct. J Biol Chem 1996; 271(12): 7177-7186. 33. Wood RD, Shivji MK: Which DNA polymerase are used for DNA repair in eucaryotes? Carcinogenesis 1997; 18; 605-610. 34. Kowalczykowski SC, Dixon DA, Eggleston AK et al.: Biochemistry of homologous recombination in Escherichia coll. Microbiol Rev 1994; 58(3): 401-465. 35. Kowalczykowski SC, Eggleston AK: Homologous pairing and DNA strand-exchange proteins. Annu Rev Biochem 1994; 63: 991-1043. 36. Kuzminov A: Unraveling the late stages of recombinational repair: metabolism of DNA junctions in Escherichia coli. Bioessays 1996; 18(9): 757-765. 37. Roca AI, Cox MM: RecA protein: structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol 1997; 56: 129-223. 38. Dixon DA, Kowalczykowski SC: The recombination hotspot chi is a regulatory sequence that acts by attenuating the nuclease activity of the E. coli RecBCD enzyme. Cell 1993; 73(1): 87-96. 39. Anderson DG, Kowalczykowski SC: The recombination hot spot chi is a regulatory element that switches. Genes Dev 1997; 11(5): 571-581. 40. Stahl FW, Stahl MM: Recombination pathway specificity of Chi. Genetics 1977; 86(4): 715-725. 41. Smith KC, Wang TV, Sharma RC: RecA-dependent DNA repair in UV-irradiated Escherichia coli. J Photochem Photobiol B 1987; 1(1): 1-11. 42. Umezu K, Kolodner RD: Protein interactions in genetic recombination in Escherichia coli. Interactions involving RecO and RecR overcome the inhibition of RecA by single-stranded DNA-binding protein. J Biol Chem 1994; 269(47): 30005-30013. 43. West SC: Processing of recombination intermediates by the RuvABC proteins. Annu Rev Genet 1997; 31: 213-244. 44. Grilley M, Griffith J, Modrich P: Bidirectional excision in methyl-directed mismatch repair. J Biol Chem 1993; 268 (16): 11830-11837. 45. Allen DJ, Makhov A, Grilley M et al.: MutS mediates heteroduplex loop formation by a translocation mechanism. EMBO J 1997; 16: 4467-4476. 46. Mechanic LE, Frankel BL, Matson SW: Escherichia coli MutL loads DNA helicase II onto DNA. J Biol Chem 2000; 275: 38337-38346. 47. Ni TT, Marsischky GT, Kolodner RD: MSH2 and MSH6 are required for removal of adenine misincorporated opposite 8-oxo-guanine in S. cerevisiae. Mol Cell 1999: 4: 439-444. 48. Acharya S, Wilson T, Gradia S et al.: hMSH2 forms specific mispair-binding complexes with hMSH3 and hMSH6. Proc. Natl. Acad. Sci. USA 1996; 93: 13629-13634. 49. Kolodner RD, Marsischky GT: Eukaryotic DNA mismatch repair. Curr Opin Genet Dev 1999; 9: 89-96. 50. Johnson KA, Mierzwa ML, Fink SP et al.: MutS recognition of exocyclic DNA adducts that are endogenous products of lipid oxidation J Biol Chem 1999; 38: 27112-27118. 51. Kowalczyk P: The influence of exocyclic DNA adducts in bacterial and mammalian genome instability. New Medicine 2012; 3: 68-73. 52. Kowalczyk P: M13mp18 phage model as a tools of research mutagenie and cytotoxic biological and environmental compounds. New Medicine 2012; 4: 116-121.
