© Borgis - Postępy Nauk Medycznych 4/2013, s. 316-325
*Michał Wąsowski, Ewa Marcinowska-Suchowierska
Otyłość a nowotwory
The link between obesity and cancer
Internal, Family Medicine and Metabolic Bone Disease Department, Medical Centre of Postgraduate Education, Warsaw
Head of Department: prof. Ewa Marcinowska-Suchowierska, MD, PhD
Streszczenie
Szereg badań klinicznych i epidemiologicznych pokazuje związek pomiędzy otyłością a nowotworami jelita grubego, rakiem piersi, endometrium, rakiem nerki czy przełyku. Dane te, jak i rosnące rozpowszechnienie otyłości sugerują, że przyrost wagi może być jednym z ważniejszych modyfikowalnych czynników wpływających na występowanie nowotworów (1).
Część powiązań pomiędzy otyłością i częstszym występowaniem niektórych typów nowotworów w tej grupie chorych może być wyjaśniona poprzez pewne zmiany funkcjonalne lub wpływ hormonów związanych z otyłością. Ważną rolę w procesie kancerogenezy odgrywają omówione w poniższym tekście mechanizmy biologiczne.
Zdecydowana większość danych sugeruje, że kombinacja czynników wydzielanych przez adipocyty (m.in. wzrost stężenia leptyny, obniżone stężenie adiponektyny i wzrost wydzielania cytokin zapalnych) oraz wpływ wtórnych do otyłości hiperinsulinemii i hiperlipidemii prowadzą do wzrostu zachorowań na raka (2).
Summary
Clinical and epidemiological prospective studies show a significant association between obesity and several cancers e.g. cancers of the colon, female breast (postmenopausal), endometrium, kidney (renal cell), and esophagus (adenocarcinoma). These data, and the year by year rising, worldwide trend in obesity, suggest that weight gain may be the largest avoidable cause of cancer in nonsmokers (1).
Although some of these associations can be explained by changes in the constitution of the human body or hormones associated with obesity, there are increases in a large variety of tumor types, suggesting that fundamental biological mechanisms may underlie these links.
The overwhelming majority of the data suggests that a combination of factors secreted by the adipocyte (increased leptin, decreased adiponectin and increased inflammatory cytokine secretion) with contributions from the secondary effects of obesity (such as hyperinsulinaemia and hyperlipidemia) lead to increased incidence of cancer (2).

OBESITY AND CANCER
Human simple obesity is defined as an imbalance of elevated caloric intake and a relative lack of physical activity. Increased mass of the adipose tissue is associated with metabolic changes described as metabolic syndrome – characterized by abdominal obesity, reduced high-density lipoprotein (HDL) cholesterol levels, increased levels of triglycerides, hypertension and insulin resistance.
A relationship between excess body weight and mortality from all causes and from cardiovascular disease has been well-established in epidemiological studies (3-9).
The adverse metabolic effects of excess body fat are known to accelerate atherogenesis and increase the risk of coronary heart disease, stroke, and early death. Obesity could also influence the growth of cancers. The relationship between obesity and cancer risk has received less attention than its cardiovascular effects. Overweight women have increased risk of endometrial cancer and breast cancer after menopause (due to increased levels of circulating estrogen). Large prospective studies show a significant association between obesity and several cancers. Obesity can play a prominent role in the incidence and progression of cancers (1).
The International Agency for Research on Cancer (IARC) in 2002 concluded that there is sufficient evidence in humans for a cancer-preventive effect of weight gain avoidance (10). Accumulating data suggests that increased adiposity may increase incidence and/or death rates from a wide variety of human cancers, including colon and rectum, esophagus (adenocarcinoma), kidney (renal cell carcinoma), pancreas, gallbladder, ovary, cervix, female breast (postmenopausal), liver, prostate, and certain hematopoietic cancers (tab. 1). With regard to premenopausal breast cancer, the report concluded that available evidence on the weight gain avoidance has no benefit as a cancer-preventive factor. These data, and the rising worldwide trend in obesity, suggest that overeating may be the largest avoidable cause of cancer in nonsmokers. Few obese people are successful in long-term weight reduction, and thus there is little direct evidence regarding the impact of weight reduction on cancer risk.
For all other sites, IARC characterized the evidence for a cancer preventive effect of avoidance of weight gain as inadequate in humans for a cancer-preventive effect of intentional weight loss for any cancer site.
Table 1. The relative risk per 5 kg per m2 increase in body mass index is reported for each site and sex.
| Cancer type | Men (95% CI) | Women (95% CI) |
| Breast | ND | 1.12 (1.08-1.16) |
| Colon | 1.24 (1.20-1.28) | 1.09 (1.05-1.13) |
| Endometrial | NA | 1.59 (1.50-1.68) |
| Oesophageal | 1.52 (1.33-1.74) | 1.51 (1.31-1.74) |
| Kidney | 1.24 (1.15-1.34) | 1.34 (1.25-1.43) |
| Leukaemia | 1.08 (1.02-1.14) | 1.17 (1.04-1.32) |
| Melanoma | 1.17 (1.05-1.30) | 0.96 (0.92-1.01) |
| Myeloma | 1.11 (1.05-1.18) | 1.11 (1.07-1.15) |
| Non-Hodgkin’s lymphoma | 1.06 (1.03-1.09) | 1.07 (1.00-1.14) |
| Pancreatic | 1.07 (0.93-1.23) | 1.12 (1.02-1.22) |
| Prostate | 1.03 (1.00-1.07) | NA |
| Rectal | 1.09 (1.06-1.12) | 1.02 (1.00-1.05) |
| Thyroid | 1.33 (1.04-1.70) | 1.14 (1.06-1.23) |
CI – confidence interval; NA – not applicable; ND – not determined.
Relative risks are taken from a meta-analysis of data as reported in Renehan et al. (11) and Roberts et al. (12).
The evidence
There is sufficient evidence in experimental animals for a cancer-preventive effect of weight gain avoidance by diet restriction, based on studies of spontaneous or chemically induced cancers of the mammary gland, liver, pituitary gland (adenoma), pancreas, for chemically induced cancers of the colon, skin (non melanoma), and prostate, and for spontaneous and genetically induced lymphoma. An association between obesity and cancer at many sites is consistent with animal studies showing that diet restriction decreases spontaneous and carcinogen-induced tumor incidence, multiplicity and size (13-15).
Obesity does not appear to have the same effect on all types of cancers, nor to affect cancer risk in both sexes. One study found that obesity increases the risk of dying from all cancers by about 52% in men, but nearly doubles the risk of dying from any type of cancer in women (16). For some cancers, such as liver cancer, obesity was linked to about a five-fold increased risk of cancer mortality in men and women together.
The association between obesity and colon cancer mortality is not equally strong in both sexes, perhaps because body mass index (BMI) is a better measure of abdominal fat in men than women, or because of hormonal factors that are protective. Obesity-related breast cancer risk also varies by menopausal status. Increasing BMI levels are linked to a lower incidence of breast cancer in premenopausal women, but a greater incidence of breast cancer in postmenopausal women.
The influence of obesity on prostate cancer risk also varies. Although obesity is associated with a lower incidence of prostate cancer, studies suggest that obesity is linked to a greater risk of being diagnosed with a more aggressive form of prostate cancer, and studies have shown that obesity increases the risk of dying from prostate cancer. Growing evidence also indicates that obesity during childhood can increase the risk of childhood cancers, such as leukemia, and young-onset brain tumors.
HOW OBESITY CAN INCREASE CANCER RISK – THE MOLECULAR MECHANISMS
Obesity is strongly associated with changes in the physiological function of adipose tissue. These processes lead to insulin resistance, chronic inflammation, and altered secretion of adipokines. Adipose tissue plays an active role in endocrine signaling to the rest of the body. It has been shown in many studies that adipose tissue secretes molecules into the bloodstream, which signal to other metabolic organs or to the brain to coordinate responses to altered metabolic demands. These molecules – adipokines, can be secreted both from the adipocyte fraction and from the stromal-vascular fraction. Some of these adipokines have a great role in modulating the risk of cancer development. The most likely contributors from the adipose tissue itself are the adipokines – leptin, adiponectin and pro-inflammatory molecules (2).
Several of these factors, such as insulin resistance, increased levels of leptin, plasminogen activator inhibitor-1, and endogenous sex steroids, decreased levels of adiponectin, and chronic inflammation, are involved in carcinogenesis and cancer progression (17).
The variability in how obesity affects the incidence, progression, or mortality of various cancers suggests that these effects derive from multiple mechanisms, which animal research supports.
Fat tissue, by producing hormones and growth factors, and by fostering inflammation, could directly fuel the growth of tumors, thereby affecting cancer incidence, progression, recurrence, and survival rates. All of these factors promote tumor initiation and growth. So, the possible mechanisms for cancerogenesis in obese include altered carcinogen metabolism, decreased oxidative DNA damage, greater DNA repair capacity (10), and a reduction of IGF-1 levels in diet restricted animals (13).
The imbalance in energy is caused by an excess of nutrients and it leads to oxidative stress and fatty acids metabolism abnormalities. It is conducive to inflammation and insulin resistance. This results in a number of processes that underlie cancer initiation and promotion, including DNA damage, cell division and migration, delayed cell death, an increase in blood vessel formation. The accelerated metabolism of fatty acids that occurs in obese individuals might increase DNA damage due to oxidation. In some evidence this DNA damage triggers a malignant transformation. The AKT-mTOR pathway is activated in obese animals, induces tumor cell growth and staves off tumor cell death. It has been implicated in a number of cancers and it is linked to an increased risk of developing a cancer, as well as to the progression of many cancers (fig. 1).
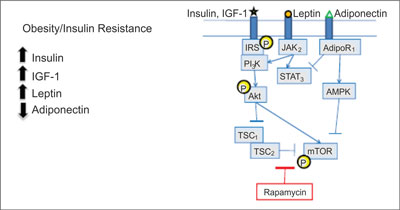
Fig. 1. Converging signaling pathways (18).
AdipoR – adiponectin receptor; AMPK – 5´adenosine monophosphateactivated protein kinase; IGF – insulin-like growth factor; IRS – insulin receptor substrate, JAK – Janus kinases; mTOR – mammalian target of rapamycin; P – phosphorylated; PI3K – phosphoinositide 3-kinase; STAT – signal transducer and activator of transcription; TSC – tuberous sclerosis protein.
Cancer is a disorder with abnormal regulation of the growth and survival of cells. Fat cells generate many hormones, growth factors, and cytokines that can disrupt regulation of cell growth and survival. These molecular factors were estrogen, insulin, insulin like growth factor 1 (IGF-1), leptin, adiponectin, and adipokinase, as well as several mediators of inflammation (fig. 2).
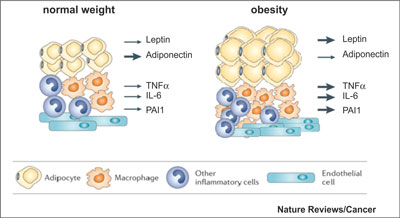
Fig. 2. Changes in adipose tissue in obesity (2).
Estrogen
The postmenopausal women are about 70% of all breast cancer patients. Most of the estrogen produced in postmenopausal women is derived from fat tissue via aromatase, the enzyme converting adrenal androgens into estrogen. The more fat tissue there is, the greater the levels of estrogen produced and in circulation. Such estrogen can fuel the growth of estrogen receptor – positive breast cancers. Studies show that mice made obese by being fed a high-fat diet and then inoculated with breast cancer cells had significantly greater tumor growth rates than mice similarly inoculated, but fed a normal diet (19). When inoculated obese mice were given an aromatase inhibitor, the tumor growth rate was markedly inhibited. Clinical studies confirm that circulating estradiol levels are linked to risk of recurrence of breast cancer. It does not explain the association of obesity with premenopausal breast cancer outcomes or with estrogen receptor – negative breast cancer outcomes (20). This mechanism also does not explain why estrogen is linked to both pre- and postmenopausal endometrial cancer risk (21).
Insulin
Another factor making obese women more susceptible to breast cancer recurrence and death is higher than normal insulin level, usually linked to obesity. BMI increase correlate closely with increases in fasting insulin levels in the nondiabetic population. Greater levels of insulin are linked to an increased risk of distant recurrence and death in breast cancer patients (22, 23).
The fetal version of the insulin receptor is overexpressed in breast cancer cells and can combine with itself or with IGF-1 to turn on the PI3K or Ras/Raf signaling pathways known to foster the growth of several types of cancers. In early-stage breast cancer women, total expression of the fetal insulin receptor is linked to worse survival rates, as is activation of the receptor by IGF-1. Insulin effects on breast cancer prognosis often are not apparent 5 years after diagnosis, suggesting that insulin may be an early mediator of the prognostic effects of obesity in breast cancer, other factors are going to be important later on. Leptin may be one of these factors. Higher levels of leptin are linked to an increased risk of distant recurrence and death from breast cancer – an effect that persists beyond 5 years postdiagnosis (24).
OTHER MOLECULAR MECHANISMS
A number of other hormones, enzymes, and growth factors, that govern cellular energy balance and growth are thought to play a role in increasing cancer risk in obese individuals, including IGF, AMP kinase, leptin, adiponectin, inflammatory cytokines.
IGFs (insulin-like growth factors)
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Calle EE, Thyn MJ: Obesity and cancer. Oncogene 2004; 23: 6365-6378.
2. Khandekar MJ, Cohen P, Spiegelman B et al.: Molecular mechanisms of cancer development in obesity. Nature Reviews Cancer 2011; 11: 886-895.
3. Manson JE, Willett WC, Stampfer MJ et al.: Body weight and mortality among women. N Engl J Med 1995; 333: 677-685.
4. Willett WC, Manson JE, Stampfer MJ et al.: Weight, weight change, and coronary heart disease in women. Risk within the ‘normal’ weight range. JAMA 1995; 273: 461-465.
5. Lindsted KD, Singh PD: Body mass and 26 y risk of mortality among men who never smoked: a re-analysis among men from the Adventist Mortality Study. Int J Obes 1998; 22: 544-548.
6. Stevens J, Cai J, Wood J: Age, body-mass index, and mortality: authors reply. New England Journal of Medicine 1998; 338: 1159.
7. Calle E, Thun M, Petrelli J et al.: Body-mass index and mortality in a prospective cohort of U.S. adults. N Engl J Med 1999; 341: 1097-1105.
8. National Institutes of Health and National Heart Lung and Blood Institute. Obes Res 1998; 6: 51S-209S.
9. National Task Force on the Prevention and Treatment of Obesity (2000). Arch Intern Med 2000; 160: 898-904.
10. IARC (2002). IARC Handbooks of Cancer Prevention. Weight Control and Physical Activity. International Agency for Research on Cancer: Lyon.
11. Renehan AG, Tyson M, Egger M et al.: Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. Lancet 2008; 371: 569-578.
12. Roberts DL, Dive C, Renehan AG: Biological mechanisms linking obesity and cancer risk: new perspectives. Annu Rev Med 2010; 61: 301-316.
13. Dunn SE, Kari FW, French J, Leininger JR et al.: Dietary restriction reduces insulin-like growth factor I levels, which modulates apoptosis, cell proliferation, and tumor progression in p53-deficient mice. Cancer Res 1997; 57: 4667-4672.
14. Hursting S, Perkins S, Brown C et al.: Calorie restriction induces a p53-independent delay of spontaneous carcinogenesis in p53-deficient and wild-type mice. Cancer Res 1997; 57: 2843-2846.
15. Kritchevsky D: Caloric restriction and experimental carcinogenesis. Toxicol Sci 1999; 52: 13-16.
16. Calle E, Rodriguez C, Walker-Thurmond K et al.: Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 2003; 348: 1625-1638.
17. van Kruijsdijk RC, van der Wall E, Visseren FL: Obesity and cancer: the role of dysfunctional adipose tissue. Cancer Epidemiol Biomarkers Prev 2009 Oct; 18(10): 2569-2578.
18. The role of obesity in Cancer Survival and Recurrence: Workshop Summary. IOM 2012. Washington DC: The National Academies Press.
19. Sabnis AJ, Cheung LS, Dail M et al.: Oncogenic Kras initiates leukemia in hematopoietic stem cells. PLoS Biol 2009; 7: e59. doi: 10.1371/journal.pbio.1000059.
20. Pierce JP, Natarajan L, Caan BJ et al.: Influence of a diet very high in vegetables, fruit, and fiber and low in fat on prognosis following treatment for breast cancer: the Women’s Healthy Eating and Living (WHEL) randomized trial. JAMA 2007; 298(3): 289-298.
21. Hamilton-Reeves JM, Rebello SA, William T et al.: Isoflavone-Rich Soy Protein Isolate Suppresses Androgen Receptor Expression without Altering Estrogen Receptor-β Expression or Serum Hormonal Profiles in Men at High Risk of Prostate Cancer. J Nutr 2007; 137(7): 1769-1775.
22. Gallagher EJ, LeRoith D: Minireview: IGF, insulin, and cancer. Endocrinology 2011; 152: 2546-2551.
23. Goodwin PJ, Ennis M, Pritchard K et al.: Fasting insulin and outcome in early-stage breast cancer: results of a prospective cohort study. J Clin Oncol 2002; 20(1): 42-51.
24. Goodwin PJ, Ennis M, Pritchard K et al.: Insulin- and obesity--related variables in early-stage breast cancer: correlations and time course of prognostic associations. J Clin Oncol 2012; 30(2): 164-171.
25. Le Roith D: Insulin-Like Growth Factors. N Engl J Med 1997; 336: 633-640.
26. Pollak M: Insulin and insulin-like growth factor signalling in neoplasia. Nature Rev Cancer 2008; 8: 915-928.
27. Boni-Schnetzler M, Schmid C, Meier PJ et al.: Insulin regulates insulin-like growth factor I mRNA in rat hepatocytes. Am J Physiol 1991; 260: E846-851.
28. Zhang L et al.: Gene expression profiles in normal and cancer cells. Science 1997; 276: 1268-1272.
29. Frystyk J, Skjaerbaek C, Vestbo E et al.: Circulating levels of free insulin-like growth factors in obese subjects: the impact of type 2 diabetes. Diabetes Metab Res Rev 1999; 15: 314-322.
30. Frystyk J, Brick DJ, Gerweck et al.: Bioactive insulin-like growth factor-I in obesity. J Clin Endocrinol Metab 2009; 94: 3093-3097.
31. Zhang Y, Proenca R, Maffei M et al.: Positional cloning of the mouse obese gene and its human homologue. Nature 1994; 372: 425-432.
32. Cohen P, Zhao C, Cai X et al.: Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest 2001; 108: 1113-1121.
33. Snoussi K, Strosberg AD, Noureddine B et al.: Leptin and leptin receptor polymorphisms are associated with increased risk and poor prognosis of breast carcinoma. BMC Cancer 2006; 6: 38.
34. Howard JM, Pidgeon GP, Reynolds JV: Leptin and gastro-intestinal malignancies. Obes Rev 2010; 11: 863-874.
35. Jarde T, Perrier S, Vasson MP et al.: Molecular mechanisms of leptin and adiponectin in breast cancer. Eur J Cancer 2011; 47: 33-43.
36. Tworoger SS, Eliassen AH, Kelesidis T et al.: Plasma adiponectin concentrations and risk of incident breast cancer. J Clin Endocrinol Metab2007; 92: 1510-1516.
37. Dal Maso L, Augustin LSA, Karalis A et al.: Circulating adiponectin and endometrial cancer risk. J Clin Endocrinol Metab 2004; 89: 1160-1163.
38. Cust AE, Kaaks R, Friedenreich C et al.: Plasma adiponectin levels and endometrial cancer risk in pre- and postmenopausal women. J Clin Endocrinol Metab 2007; 92: 255-263.
39. Soliman PT, Cui X, Zhang Q et al.: Circulating adiponectin levels and risk of endometrial cancer: the prospective Nurses’ Health Study. Am J Obstet Gynecol 2011; 204: 167 e1-e5.
40. Bub JD, Miyazaki T, Iwamoto Y: Adiponectin as a growth inhibitor in prostate cancer cells. Biochem Biophys Res Commun 2006; 340: 1158-1166.
41. Kim AY, Yun SL, Kang HK et al.: Adiponectin represses colon cancer cell proliferation via AdipoR1- and-R2-mediated AMPK activation. Mol Endocrinol 2010; 24: 1441-1452.
42. Lam JB, Chow KHM, Xu A et al.: Adiponectin haploinsufficiency promotes mammary tumor development in MMTV-PyVT mice by modulation of phosphatase and tensin homolog activities. PLoS One4 2009; e4968.
43. Fogarty S, Hardie DG: Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochem Biophys Acta 2010; 1804: 581-591.
44. Sharma D, Wang J, Fu PP et al.: Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology 2010; 52: 1713-1722.
45. Sun Y, Lodish HF: Adiponectin deficiency promotes tumor growth in mice by reducing macrophage infiltration. PLoS One5 2010; e11987.
46. Chen J: Multiple signal pathways in obesity-associated cancer. Obes Rev 2011; 12(12): 1063-1070.
47. Hotamisligil GS, Shargill NS, Spiegelman BM: Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993; 259: 87-91.
48. Hotamisligil GS, Arner P, Caro JF et al.: Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 1995; 95: 2409-2415.
49. Fried SK, Bunkin DA, Greenberg AS: Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab 1998; 83: 847-850.
50. Sawdey MS, Loskutoff DJ: Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha, and transforming growth factor-beta. J Clin Invest 1991; 88: 1346-1353.
51. Park EJ, Lee JH, Yu GY et al.: Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell2010; 140: 197-208.
52. Bredel M, Scholtens DM, Yadav A et al.: NFKBIA Deletion in Glioblastomas. N Engl J Med 2011; 364: 627-637.
53. Calado DP, Zhang B, Srinivasan L et al.: Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell 2010; 18: 580-589.
54. Wang W, Abbruzzese JL, Evans DB et al.: The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin Cancer Res 1999; 5: 119-127.
55. Pikarsky E, Porat RM, Stein I et al.: NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004; 431: 461-466.
56. Kern PA, Ranganathan S, Li C et al.: Adipose tissue tumor necrosis factor and interleukin-6 expression in human obesity and insulin resistance. Am J Physiol Endocrinol Metab 2001; 280: E745-751.
57. Mohamed-Ali V, Goodrick S, Rawesh A et al.: Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab 1997; 82: 4196-4200.
58. Bromberg JF, Wrzeszczynska MH, Devgan G et al.: Stat3 as an oncogene. Cell 1999; 98: 295-303.
59. Vaisse C, Halaas JL, Horvath CM et al.: Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nature Genet 1996; 14: 95-97.
60. Giovannucci E, Colditz GA, Stampfer MJ et al.: Physical activity, obesity, and risk of colorectal adenoma in women (United States).Cancer Causes Control 1996; 7: 253-263.
61. Dignam JJ, Polite BN, Yothers G et al.: Body mass index and outcomes in patients who receive adjuvant chemotherapy for colon cancer. J Natl Cancer Inst 2006; 98(22): 1647-1654.
62. Giovannucci E, Ascherio A, Rimm E et al.: Physical activity, obesity, and risk for colon cancer and adenoma in men. Ann Intern Med 1995; 122: 327-334.
63. Calle EE, Miracle-McMahill HL, Thun MJ et al.: Estrogen replacement therapy and risk of fatal colon cancer in a prospective cohort of postmenopausal women J Natl Cancer Inst 1995; 87: 517-523.
64. Rossouw JE, Anderson GL, Prentice RL et al.: Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288(3): 321-33.
65. Sandhu M, Dunger D and Giovannucci E: Insulin, insulin-like growth factor-I (IGF-I), IGF binding proteins, their biologic interactions, and colorectal cancer. J Natl Canc Inst 2002; 94: 972-980.
66. Komninou D, Ayonote A, Richie JJ et al.: Insulin resistance and its contribution to colon carcinogenesis. Exp Biol Med 2003; 228: 396-405.
67. Macaulay VM: Insulin-like growth factors and cancer. Br J Cancer 1992; 65(3): 311-320.
68. LeRoith D: Seminars in medicine of the Beth Israel Deaconess Medical Center. Insulin-like growth factors. N Engl J Med 1997 Feb 27; 336(9): 633-640.
69. Schoen R, Tangen C, Kuller L et al.: Increased blood glucose and insulin, body size, and incident colorectal cancer. J Natl Canc Inst 1999; 91: 1147-1154.
70. Kaaks R, Van Noord PA, DenTonkelaar I et al.: Breast-cancer incidence in relation to height, weight and body-fat distribution in the Dutch “DOM” cohort. Int J Cancer 1998; 76: 647-651.
71. Ma J, Pollak M, Giovannucci E et al.: Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer Inst 1999; 91: 620-625.
72. Giovannucci E, Pollak MN, Platz E et al.: Prospective study of plasma insulin-like growth factor-1 and binding protein-3 and risk of colorectal neoplasia in women. Cancer Epidemiol Biomark Prev 2000; 9: 345-349.
73. Manousos O, Souglakos J, Bosetti C et al.: IGF-I and IGF-II in relation to colorectal cancer. Int J Cancer 1999; 83: 15-17.
74. Renehan A, Jones P, Potten C et al.: Elevated serum insulin-like growth factor (IGF)-II and IGF binding protein-2 in patients with colorectal cancer. Br J Cancer 2000; 83: 1344-1350.
75. Potischman N, Swanson C, Siiteri P et al.: Reversal of relation between body mass and endogenous estrogen concentrations with menopausal status. J Natl Canc Inst 1996; 88: 756-758.
76. Hunter DJ, Willett WC: Diet, body size, and breast cancer. Epidemiol Rev 1993; 15: 110-132.
77. Ballard-Barbash R, Swanson C: Body weight: estimation of risk for breast and endometrial cancers. Am J Clin Nutr 1996; 63(suppl): 437S-331S.
78. Trentham-Dietz A, Newcomb PA, Storer BE et al.: Body size and risk of breast cancer. Am J Epidemiol 1997; 145: 1011-1019.
79. Galanis DJ, Kolonel LN, Lee J et al.: Anthropometric predic- tors of breast cancer incidence and survival in a multi-ethnic cohort of female residents of Hawaii, United States. Cancer Causes Control 1998; 9(2): 217-224.
80. Folsom AR, Kaye SA, Sellers TA et al.: Body Fat Distribution and 5-Year Risk of Death in Older Women. JAMA 1993; 269: 483-487.
81. Kaaks R, Van Noord PA, DenTonkelaar I et al.: Breast cancer incidence in relation to height, body-fat distribution in the Dutch. ‘DOM’ cohort. Int J Cancer 1998; 76: 647-651.
82. Folsom AR, Kaye SA, Prineas RJ et al.: Increased incidence of carcinoma of the breast associated with abdominal adiposity in postmenopausal women. Am J Epidemiol 1990; 131: 794-803.
83. Protani M, Coory M, Martin JH: Effect of obesity on survival of women with breast cancer: systematic review and meta-analysis. Breast Cancer Res Treat 2010; 123(3): 627-635.
84. Key T, Appleby P, Barnes I et al.: Endogenous sex hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. The Endogenous Hormones and Breast Cancer Collaborative Group. J Natl Canc Inst 2002; 94: 606-616.
85. Cao Y, Ma J: Body mass index, prostate cancer-specific mortality, and biochemical recurrence: a systematic review and meta-analysis. Cancer Prev Res (Phila) 2011 Apr; 4(4): 486-501.
86. Freedland SJ, Grubb KA, Yiu SK et al.: Obesity and risk of biochemical progression following radical prostatectomy at a tertiary care referral center. J Urol 2005; 174(3): 919-922.
87. Ma J, Li H, Giovannucci E et al.: Prediagnostic body-mass index, plasma C-peptide concentration, and prostate cancer-specific mortality in men with prostate cancer: A long-term survival analysis. Lancet Oncol 2008; 9(11): 1039-1047.
88. IOM. 2012c. Alliances for obesity prevention: Finding common ground: Workshop summary. Washington, DC: The National Academies Press.
