© Borgis - Medycyna Rodzinna 4/2004, s. 184-186
Jolanta Kłopocka
Dziedziczna sferocytoza – problemy diagnostyczne w świetle najnowszych badań
Hereditary spherocytosis – diagnostic problems in view of new research
z Zakładu Diagnostyki Laboratoryjnej CMPK w Warszawie
Kierownik Zakładu: prof. dr hab. med. Dagna Bobilewicz
Obecnie: Zakład Biofizyki i Biomatematyki CMKP
Summary
Hereditary spherocytosis (H.S) is a heterogeneous syndrome in terms of clinical severity, inheritance, red blood cells morphology as well as in molecular defects. The main problems connected with diagnosis of H.S. are discussed.

Dziedziczna sferocytoza (H.S.) jest najczęściej występującą wrodzoną niedokrwistością hemolityczną w populacji północnoeuropejskiej. W północnej Europie i USA występuje z częstością 1 na 5000 mieszkańców (32). Jest to zaburzenie dziedziczne charakteryzujące się obecnością sferocytów w rozmazach krwi obwodowej. Głównym defektem krwinek czerwonych w H.S. jest utrata prawidłowej powierzchni błony komórkowej, co jest przyczyną ich sferycznego kształtu, jak i zmniejszonej odkształcalności. Niszczenie w śledzionie tych nieprawidłowych komórek jest podstawową przyczyną hemolizy u pacjentów z H.S.
Cytoszkielet erytrocyta decydujący o jego charakterystycznym kształcie jest utworzony pod błoną komórkową z białek: spektryny, ankyryny, białka prążka 3 i białka prążka 4,2 (wybarwione prążki obserwowane na żelu po elektroforetycznym rozdziale białek erytrocyta) (ryc. 1). U pacjentów z H.S. dochodzi do niedoborów białek błony komórkowej krwinek czerwonych, głównie spektryny, a także łącznego niedoboru spektryny i ankyryny, białka 3 oraz w niewielkim stopniu białka 4,2 (10, 32, 33, 43, 44). Badania przeprowadzone między innymi przez Palka i Jarolima dotyczące tych białek u pacjentów z H.S. wykazują ich jakościowe i/lub ilościowe różnice (10, 12, 32, 38, 39, 46).
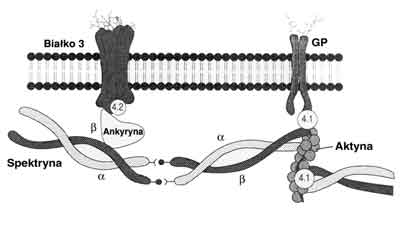
Ryc. 1. Błona komórkowa erytrocyta. Schematycznie przedstawione białka szkieletowe błony komórkowej erytrocyta: a i b-spektryna, ankyryna, białko prążka 3, białko prążka 4,1 i 4,2, aktyna, GP-glikoforyna.
Łączny niedobór spektryny i ankyryny można wytłumaczyć tym, że zmniejszona synteza ankyryny oraz zmniejszony jej udział w budowie błony komórkowej prowadzi do zmniejszonego wiązania spektryny na ankyrynie (21, 32). Erytrocytarna spektryna utrzymuje kształt komórki, reguluje ruchliwość integralnych białek błony komórkowej i stanowi strukturalne podparcie dla podwójnej warstwy lipidów.
Spektryna złożona jest z dwóch podjednostek a- i b-spektryny. Pomimo pewnego podobieństwa różnią się one pod względem strukturalnym, a także kodowane są przez oddzielne geny (6, 9, 23, 32, 45). Geny dla a - i b- spektryny ludzkich erytrocytów znajdują się odpowiednio na chromosomie 1 i 14. Gen dla ankyryny natomiast na chromosomie 8 (5, 10, 24), dla białka 3 na chromosomie 17, a dla białka 4,2 na 15. (32).
Jak wykazały badania, niedobór spektryny u pacjentów z H.S. dziedziczy się w sposób dominujący, jak i recesywny (19, 27, 44). Niedobór a-spektryny zależy od mutacji genu a-spektryny i związany jest z recesywnym sposobem dziedziczenia. W obu znanych wariantach Praga i Lepra stwierdzono mutację zmiany fazy odczytu (10).
Niedobór b-spektryny natomiast zależy od mutacji genu b-spektryny i związany jest głównie z dominującym sposobem dziedziczenia (1, 2, 11, 13, 32). Wyjątek stanowi wariant Birmingham, w którym niedobór b-spektryny jest dziedziczony w sposób recesywny.
W wariancie Atlanta stwierdzono mutację zmiany sensu (182 Trp – Gly), w wariancie Philadelphia mutację zmiany fazy odczytu związaną z insercją, a w wariancie Ostrava z delecją. Stwierdzono również kilka mutacji typu nonsens, jak Alger, Baltimore, Tabor (1, 2, 10, 11).
Przeprowadzone badania wykazały również liczne mutacje genu ankyryny (7, 15, 18, 29-31, 37), między innymi, takie jak mutację zmiany fazy odczytu związaną z insercją – wariant Einbeck lub delecją – wariant Napoli II, typu nonsens wariant Rakownik.
Niedobór ankyryny dziedziczy się głównie w sposób dominujący (10). W ten sam sposób dziedziczy się niedobór białka 3 i jest to związane z mutacją genu białka 3 (3, 4, 16,17, 25, 26, 34, 35, 36, 42). W wariancie Praga opisano mutację zmiany fazy odczytu związaną z insercją (8, 32). W innych wariantach występowała między innymi delecja – wariant Every, a także mutacja zmiany sensu Montefiore (40 Glu – Lys).
Niedobór białka 4,2 jest powszechny u pacjentów z H.S. w Japonii. Zależy on od mutacji genu białka 4,2 i dziedziczy się w sposób recesywny (8, 14, 22, 28, 41, 47). U niektórych pacjentów między innymi stwierdzono mutację zmiany sensu (142 Ala – Thr).
H.S. jest heterogenna pod względem przebiegu klinicznego, sposobu dziedziczenia, morfologii krwinek czerwonych, jak również występowania defektów molekularnych i ze względu na to stanowi ona od lat problem diagnostyczny. U pacjentów z ostrym przebiegiem H.S. we krwi obwodowej stwierdza się mikrosferocyty. W H.S. z łagodnym przebiegiem klinicznym, gdzie utrata powierzchni błony komórkowej jest mała, krwinki czerwone mogą zachować kształt dwuwklęsły. W konsekwencji może to prowadzić do złego rozpoznania.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Basseres D.S., et al.: Blood, 1997, 90 Suppl.: 46 (Abstract). 2.Basseres D.S., et al.: Blood, 1998, 91, 368. 3.Bianachi P., et al.: Blood, 1995, 468a (Abstr. Suppl 1). 4.Bracher N., et al.: Br. J. Haematol., 2001, 113, 689. 5.Costa F.F., et al.: N. Engl. J. Med., 1990, 323, 1046. 6.Dhermy D., et al.: Blood Cells Mol. Dis., 1998, 24 (12) June 30, 251. 7.Eber S. W. et al.: Nat. Genet., 1996, 13, 214. 8.Gallager P.G.: Blood Cells Mol. Dis., 1997, 23, (22), Nov. 30, 417. 9.Gallager P.G., Forget B.: Sem. Hematol., 1993, 30, 4. 10.Gallager P.G., Forget B.: Blood Cells Mol. Dis.,1998, 24 (23), Dec. 15, 539. 11.Garbarz M., et al.: Br. J. Haematol., 1998, 100, 90. 12.Hanspal M., et al.: Blood, 1991, 77, 165. 13.Hassoun H., et al.: Blood, 1997, 90, 1, 398. 14.Hayette S., et al.: Blood, 1995, 85, 1, 250. 15.Hayette S., et al.: Am. J. Hematol., 1998, 58, 36. 16.Jarolim P., et al.: Blood, 1995, 85, 3,634. 17.Jenkins P. B., et al.: J. Clin. Invest., 1996, 97, 373. 18.Kanazaki A., et al.: Blood, 1997, 90, (Suppl. 1), 6B (Abstract). 19.Karan A.S., Saxena R.: Am. J. Hematol., 2002, 70, 266. 20.Kłopocka J., Jabłońska-Skwiecińska E.: Diag. Lab., 1993, 29, 3, 315. 21.Komada M., Soriano Ph.: J. Cell. Biol., 2002, 156, 337. 22.Korsgren C., et al.: Proc. Natl. Acad. Sci. U.S.A., 1991, 88, 4840. 23.Luo B.H., et al.: Eur. J. Haematol., 2002, 68, 73. 24.Lux S.E., et al.: Nature, 1990, 345, 736. 25.Miraglia del Giudice E.: Blood, 1995, (Abstr.Suppl. 1), 86, 631. 26.Miraglia del Giudice E., et al.: Br. J. Haemat., 1997, 96, 70. 27.Miragalia del Giudice., et al.: Br. J. Haematol., 2001, 112, 42. 28.Matsuda M., et al.: Hum. Mol. Genet., 1995, 4, 1187. 29.Morle L., et al.: Blood, 1996, (Abstr. Suppl. 1), 86, 467 a. 30.Morle L., et al.: Am. J. Hemat., 1997, 54, 242. 31.Ozcan R., et al.: Blood, 1997, 90 (Suppl.): 4a (Abstract). 32.Palek J., Jarolim P.: Semin. Hematol., 1993, 30, 4, 249. 33.Palek J.: Semin. Hematol., 1993, 30, 1, 1. 34.Perrota S., et al.: Blood, 1999, 93, 6, Murch. 15. 35.Perrota S., et al.: Blood, 1999, 93, 6, March 15. 36.Peters L. L., et al.: Cell, 1996, 86, 917. 37.Randon J., et al.: B. J. Haematol., 1997, 96, 500. 38.Saad S. T.: Br. J. Haemat., 1994, 88, 295. 39.Savvides P., et al.: Blood, 1993, 82, 2953. 40.Streichman S., Gescheidt Y.: Am. J. Hematol., 1998, 58, 206. 41.Takaoka Y. et al.: B. J. Haemat., 1994, 88, 527. 42.Tanner M.J.A.: Sem. Hematol., 1993, 30, 1, 34. 43.Wandersee N., et al.: Blood, 2001, 97, 543. 44.Wichterle H., et al.: Blood, 1995, 86, 468a. 45.Winkelman J.C., Forget B.G.: Blood, 1993, 81, 3173. 46.Witfield C.F.: Blood, 1991, 78, 3043. 47.Yawata Y., et al.: Int. J. Haematol., 2000, 71, 118.
