© Borgis - New Medicine 2/2009, s. 45-47
*Justyna Paprocka1, Ewa Jamroz1, Antoni Pyrkosz2, Elżbieta Marszał1
Freeman-Sheldon syndrome
1Child Neurology Department, Silesian Medical University, Katowice, Poland
2Department of Clinical and Molecular Genetics, Silesian Medical University, Katowice, Poland
Head of the Department: Prof. Elżbieta Marszał, MD, PhD
Summary
Freeman-Sheldon is a rare and underdiagnosed distal arthrogryposis syndrome. It is the most severe form and despite the striking contractures of the orofacial muscles (resulting in down-slanting palpebral fissures, prominent nasolabial folds, H-shaped dimpling of the chin, pinched lips) may be misdiagnosed as a Sheldon-Hall syndrome. The authors describe a 14-month-old boy with the phenotype of Freeman-Sheldon syndrome. Despite typical presentation, however, many physicians remain unfamiliar with the condition. Its diagnosis has important implications for both medical management and counselling.
Introduction
In the mid-1990s the classification of the most common Mendelian-inherited arthrogryposis syndromes was revised (1). Distal arthrogryposes (DAs) were classified into 10 hierarchically related disorders according to the proportion of features they shared with one another (DA1-DA10). Features shared among the DAs include a consistent pattern of distal joint (hands and feet) involvement, limited proximal joint involvement, autosomal dominant inheritance, and variable expressivity. Most FSS cases are sporadic, but there is also evidence of autosomal-dominant transmission in FSS. An autosomal recessive or X-linked recessive pattern may be observed in cases in which the parent has non-penetrant somatic mosaicism or germ-like mosaicism.
The combined prevalence of all causes of congenital contracture is 0.5-1.0%, and patients with more than one congenital contracture are found in 1/3000 births (1). These cases are often sporadic, but children are frequently found to have underlying syndrome transmitted in a Mendelian pattern (2, 3, 4).
Several DA syndromes can be caused by mutations in 4 genes that encode proteins of the troponin-tropomyosin complex of fast-twitch myofibres (mutations in TPM2, TNN12, TNNT3, MYH8) (2). No mutations in TNN12 or TNNT3 were found in patients with FSS (3). Due to phenotypic overlap between different distal arthrogryposis syndromes, a hypothesis was postulated about mutations that perturb the development and function of sarcomeres (resulting in diminished fetal movement and contractures) (4). In 2006 Toydemir et al. showed that mutations in the embryonic myosin heavy chain (MYH3) gene cause Freeman-Sheldon syndrome (2).
The most frequently seen clinical manifestations are: scoliosis, dental crowding, strabismus, severe respiratory infections, hearing loss, fractures, hernia, cryptorchidism, headaches, malignant hyperthermia, joint dislocations, severe vomiting, and arthritis/joint pain (1, 5). Children with Freeman-Sheldon syndrome are recognized by the characteristic face (”whistling mouth”). Microcephaly and mental retardation have been observed in one third of patients (6, 7).
The authors present a 14-month-old boy with the phenotype of Freeman-Sheldon syndrome.
Patient presentation
14-month-old boy (fig. 1) from the second pregnancy (first pregnancy resulted in still birth) complicated with suspicion of preterm delivery, born after a 38-week gestation period by spontaneous vaginal delivery with birth weight of 4030 g, Apgar score of 10 points. In the family history hypothyroidism in the mother was diagnosed. On the 11th day of life the child was transferred to the Neonatal Intensive Care Unit because of congenital anomalies (dysmorphy: whistling face, microstomia, broad nasal cartilages, deep set eyes, deformity of upper extremities (ulnar deviation and flexor contractures), inguinal hernia, hip dysplasia, deformity of feet (clubfoot)). MRI of the head, performed in the neonatal period, showed normal brain structures. Because of severe vomiting the patient underwent pH-metry and roentgen of the gastrointestinal tract, which confirmed gastroesophageal reflux. Two operations were performed because of inguinal hernias. During infancy frequent respiratory infections were present, including Pneumocystis carini as a pathogen. Psychomotor development was delayed – the child cannot roll over or sit unaided. Because of oral contractures supplemental feeding techniques were provided with a modified nipple with a widened opening.

Fig. 1. Patient with Freeman-Sheldon syndrome.
On admission in the physical examination the child presented with the phenotype of Freeman-Sheldon syndrome. In neurological examination head circumference was 46.5 cm (10th centile); muscle tone diminished in the head-trunk axis, and increased in the extremities, being more pronounced in the upper and on the right side. Psychomotor development was assessed at the level of 4 months.
MRI of the head (14 months) showed dysmyelination in white matter of parietooccipital lobes with cystic enlargement of the pericerebral space in the left temporal region (fig. 2-4). Fundus oculi was normal. No structural anomaly or functional disturbance was found on electro- or echocardiography. Needle electromyography of the tibial anterior muscle revealed no myopathic changes; also conduction velocity was within the normal range. In EEG tracing non-paroxysmal and paroxysmal changes were detected. During hospitalization no epileptic seizures were observed. Ultrasonography of the abdominal cavity was normal. The parents did not agree for the molecular study. The child is prepared for botulin injections.
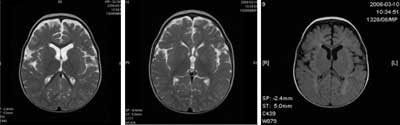
Fig. 2-4. MRI of the head (performed at the age of 11 months): T1, T2 FLAIR sequences. T2-and FLAIR-weighted MI scans show dysmyelination in white matter of parietooccipital lobes with enlargement of subarachnoid pericerebral spaces especially in the left temporal region.
Discussion
FSS was first described by Freeman and Sheldon in 1938 (5). It is generally dominant in nature although also recessive mode of inheritance is reported. Based on Stevenson´s detailed report on 32 individuals with FSS apart from contractures the most common features included severe scoliosis, strabismus, and hearing loss (Tables 1, 2).
Table 1. Contractures/limited range of motion in FSS according to Stevenson DA, Carey JC, Palumbos J et al. Clinical characteristics and natural history of Freeman-Sheldon syndrome. Pediatrics 2008; 117: 754-762.
| Location | With contracture n/N (%) |
Fingers
Neck
Wrists
Feet/ankles
Hips
Elbows
Knees |
28/28 (100)
24/27 (89)
23/27 (85)
27/27 (100)
15/26 (58)
11/25 (44)
7/27 (26)
|
Table 2. Clinical manifestation in FSS according to Stevenson DA, Carey JC, Palumbos J et al. Clinical characteristics and natural history of Freeman-Sheldon syndrome. Pediatrics 2008; 117: 754-762.
| Manifestation | With manifestation, n/N (%) |
Dental crowding
Scoliosis
Cryptorchidism
Strabismus
Frontal headaches
Severe respiratory infections
Hearing loss
Fractures
Hernia
Arthritis/joint pain
Severe vomiting
Malignant hyperthermia
Joint dislocations |
14/14 (100)
22/26 (85)
5/12 (42)
11/26 (42)
5/14 (36)
9/27 (33)
8/27 (30)
6/23 (26)
6/26 (23)
2/11 (18)
2/12 (17)
3/19 (16)
3/26 (12)
|
The diagnostic criteria for classical FSS comprise: the presence of>/2 of the major clinical manifestations of DA (ulnar deviation of the wrists and fingers, camptodactyly, hypoplastic and/or absent flexion creases, and/or overriding fingers at birth, talipes equinovarus and calcaneovalgus deformities, a vertical talus, and/or metatarsus varus) plus the presence of a small pinched mouth, prominent nasolabial folds, H-shaped dimpling of the chin.
The presented patient fulfilled criteria for FSS.
The facial contractures of FFS are similar to, although more severe than, those found in children with Sheldon-Hall syndrome (SHS).
Microcephaly and mental retardation are present in one-third of patients. The CNS abnormalities documented in FSS showed a pattern of atrophic changes particularly evident in the region of the medulla oblongata. Also earlier observed brain stem abnormalities could account for the abnormal respiratory and cough reflex that may contribute to respiratory problems described in FSS patients (8, 9). Zampino et al. described a child with FSS and cerebellar and brainstem atrophy (8). This patient presented with severe phenotype of hypertonicity, multiple episodes of pneumonia, difficulty with swallowing and poor weight gain. Kohyama and Shiiki pointed out the possibility of obstructive sleep apnoea in children with FSS (9). Aspiration pneumonia and respiratory difficulties are the main causes of early mortality (9). Long-term outcomes have been satisfactory in the majority of children.
An interesting study performed by Vimercati et al. verified the usefulness of fetal mouth length measurements. The study population comprised 341 fetuses of between 15 and 38 weeks gestation (10). They proved its effectiveness in the case of face abnormalities.
In FSS life expectancy is normal. Though respiratory challenges and complications faced by a patient with FSS can be numerous, the syndrome´s primary involvement is limited to the musculoskeletal systems, and satisfactory quality and length of life can be expected with proper care.
Piśmiennictwo
1. Stevenson DA et al.: Clinical characteristics and natural history of Freeman-Sheldon syndrome. Pediatrics 2006; 117: 754-762. 2. Toydemir RM et al.: Mutations in embryonic myosin heavy chain MYH3) cause Freeman-Sheldon syndrome and Sheldon-Hall syndrome. Nat Genet 2006; 38: 561-565. 3. Sung SS et al.: Mutations in TNNT3 cause multiple congenital contractures: a second locus for distal arthrogryposis type 2B. Am J Hum Genet 2003; 73: 212-214. 4. Sung SS, Brassington AE, Grannatt K: Mutations in genes encoding fast-twinch contractile proteins cause distal arthrogryposis syndromes. Am J Hum Genet 2003; 72: 681-690. 5. Freeman EA, Sheldon JH: Cranio-carpo-tarsal dystrophy: an undescribed congenital malformation. Arch Dis Child 1938; 13: 277-283. 6. Jangid S, Khan SA: Freeman-Sheldon syndrome. Indian Pediatr 2005; 42: 717. 7. Krakowiak PA et al.: A variant of Freeman-Sheldon syndrome maps to 11p15.5-pter. Am J Hum Genet 1997; 60: 426-32. 8. Zampino G et al.: Severe form of Freeman-Sheldon syndrome associated with brain anomalies and hearing loss. Am J Med Genet 1996; 29; 62: 293-296. 9. Kohyama J, Shiiki T: Sleep disordered breathing during REM sleep in Freeman-Sheldon syndrome. Acta Neurol Scand 2000; 102: 395-397. 10. Vimercati A et al.: Prenatal diagnosis of Freeman-Sheldon syndrome and usefulness of an ultrasound fetal lip width normogram. Prenat Diagn 2006: 26: 679-683.
