© Borgis - Postępy Nauk Medycznych 1/2008, s. 14-22
*Magdalena Walicka, Agnieszka Jasik, Marzanna Paczyńska, Michał Wąsowski, Marek Tałałaj, Ewa Marcinowska-Suchowierska
Niedobór witaminy D – problem społeczny
Vitamin D deficiency – public health problem
Klinika Medycyny Rodzinnej i Chorób Wewnętrznych
Centrum Medycznego Kształcenia Podyplomowego w Warszawie
Kierownik Kliniki: prof. dr hab. med. Ewa Marcinowska-Suchowierska
Streszczenie
Witamina D jest niezbędna do utrzymania dobrego stanu kośćca. Jej źródłem jest produkcja skórna oraz pożywienie. Zaopatrzenie organizmu ludzkiego w witaminę D zależy głównie od ekspozycji na światło słoneczne (UVB 290-315 nm). Na skórną syntezę witaminy D wpływają: pora roku, szerokość geograficzna, pora dnia, kolor skóry, wiek, użycie filtrów przeciwsłonecznych. Niedobór witaminy D wywołuje krzywicę u dzieci, a u osób dorosłych zwiększa ryzyko złamania w przebiegu osteoporozy oraz powoduje osteomalację. Niedobór witaminy D jest powszechny i dotyczy zwłaszcza ludzi starych. Zalecana dzienna podaż (RDA) 400 IU/d jest zbyt mała i wymaga zmiany. Największą barierą legislacyjną utrudniającą walkę z hipowitaminozą D stanowi fakt, że dawka 50 ?g/dobę jest górną granicą dla tej substancji w suplementach żywieniowych. Należy zatem ponownie poddać ocenie powyższe ograniczenia.
Summary
Vitamin D is essential for the maintenance of good bone health. Its sources can be skin production and diet intake. Most humans depend on sunlight exposure (UVB 290–315 nm) to satisfy their requirements for vitamin D. Season, latitude, time of day, skin pigmentation, aging, sunscreen use, all influence the cutaneous production of vitamin D3. Vitamin D deficiency not only causes rickets among children and osteomalacia among adults but also it has been associated with an increased risk for osteoporosis fracture. The prevalence of hypovitaminosis D is considerable and mainly old people are affected. A recommended dietary allowance (RDA) 400 IU per day is not enough and should by increased. Currently, the major legislative barrier to improving vitamin D status for the general public is that 50 ?g/day is the upper limit (UL) for this nutrient. The evidence for this preceding safety limit needs to be reassessed.

WPROWADZENIE
Określenie „witamina D” lub „witaminy D” obejmuje grupę związków chemicznych, o ogólnym wzorze C28H43OH, z grupy steroidów wykazujących działanie przeciwkrzywicze; spośród nich największe znaczenie praktyczne dla człowieka ma witamina D2 i witamina D3.
Witaminy D (D2 i D3) nie wykazują działania biologicznego. Są one substancjami wyjściowymi, które w organizmie ulegają cyklowi przemian prowadzącemu do wytworzenia czynnych metabolitów. Pierwszy etap to hydroksylacja witaminy D w wątrobie, w wyniku której powstaje 25-hydroksywitamina D [25(OH)D] – substancja o umiarkowanej aktywności biologicznej (przeciwkrzywiczej), stanowiąca główną formę witaminy D w krwiobiegu. Drugi etap przemian odbywa się w nerkach, gdzie zachodzi hydroksylacja w pozycji 1α prowadząc do syntezy 1,25-dihydroksywitaminy D [1,25(OH)2D], powszechnie uznanej za najbardziej aktywną postać witaminy D.
Postęp w ostatnich latach poważnie rozszerzył wiedzę zarówno o metabolizmie witaminy D, jak i mechanizmach go regulujących, a także tkankach jej działania docelowego. W wyniku tego 1,25(OH)2D uznano za hormon, a cykl przemian witaminy D objęto nazwą „system endokrynny witaminy D”.
Witamina D pełni niekwestionowaną rolę w procesach wzrostu i mineralizacji kości. Powoduje ona zwiększenie wchłaniania wapnia w przewodzie pokarmowym, jego utylizację w kościach przez wzmaganie mineralizacji nowo powstałego osteoidu, a ponadto zwiększa absorpcję wapnia i fosforanu w nerkach. Niedobór witaminy D uznany został za czynnik ryzyka rozwoju osteoporozy, ponieważ zmniejsza wytrzymałość mechaniczną szkieletu, sprzyja upadkom zwiększając ryzyko złamań (1). Celem tego opracowania jest przybliżenie przyczyn narastającego niedoboru witaminy D przez pryzmat jej metabolizmu w warunkach fizjologii oraz omówienie skutków jej niedoboru dla kośćca.
METABOLIZM WITAMINY D W WARUNKACH FIZJOLOGICZNYCH
Źródła witaminy D
Witamina D3 (cholekalcyferol) pochodzi z dwóch źródeł. Znaczna jej część znajduje się w pokarmach (ryby, jaja, wątroba zwierzęca, produkty mleczne) i ulega wchłanianiu w przewodzie pokarmowym, a część powstaje pod wpływem promieniowania ultrafioletowego działającego na znajdujący się w skórze dehydrocholesterol. Witamina D2 (ergokalcyferol) dostarczana jest do organizmu tylko doustnie w spożywanych pokarmach roślinnych oraz grzybach. Jednostką międzynarodową (IU) witaminy D jest 0,025 μg czystego kalcyferolu. Dzienna zalecana podaż (Recomended dietary allowance – RDA) witaminy D u osób dorosłych wynosi 400 IU.
Oceniając zaopatrzenie organizmu w witaminę D u osób z wyrównaną podażą doustną witaminy D2 ustalono, że około 80% znajdującej się w ustroju witaminy D pochodzi z syntezy skórnej. W krajach nie stosujących wzbogacania żywności w witaminę D procent ten jest prawdopodobnie jeszcze wyższy.
Witamina D podana doustnie wraz z pożywieniem w ponad 80% wchłania się z przewodu pokarmowego, głównie w odcinku czczym jelita cienkiego. Wchłanianie jej ułatwione jest w obecności soli i kwasów żółciowych, kwasów tłuszczowych, monoglicerydów. Zaabsorbowana witamina D przechodzi następnie do limfy, gdzie wiąże się głównie z chylomikronami (90%); wynika to z jej dobrej rozpuszczalności w tłuszczach, a zarazem wysokiego stężenia chylomikronów w limfie. Z chwilą wymieszania limfy z krwią, witamina D transportowana jest do wątroby (głównie w połączeniu z białkiem wiążącym – vitamin D binding protein [DBP], którego stężenie w krwi jest sześć razy większe niż w limfie) w celu jej hydroksylacji (1, 2).
Synteza skórna witaminy D
Endogenna synteza skórna witaminy D odbywa się zasadniczo w dwóch etapach: pierwszy – znajdujący się w skórze substrat, 7-dehydrocholesterol (7DHC), ulega przekształceniu w prowitaminę D (pre-D3) pod wpływem promieniowania ultrafioletowego (UV) o długości fali 290-320 nm (UVB), drugi – prowitamina D3 pod wpływem temperatury ciała ulega przekształceniu w witaminę D3. Witamina D3 powstająca w głębszych warstwach naskórka w pobliżu naczyń krwionośnych i limfatycznych wiązana jest całkowicie przez DBP – białko cechujące się ogromną pojemnością i siłą wiązania witaminy D.
Badania nad syntezą skórną witaminy D ujawniły istotne różnice w jej fotosyntezie zależnie od czasu ekspozycji skóry na UVB. Ustalono, że po krótkiej jednorazowej ekspozycji skóry na promieniowanie UV następuje szybka (w ciągu 15 min.) przemiana 7DHC do pre-D3, która magazynowana jest w naskórku, a następnie – pod wpływem temperatury ciała – pre-D3 ulega przekształceniu w witaminę D3 (10% w ciągu 6 h), która powoli uwalniana jest do krwiobiegu (3 dni). Przemiana taka sprzyja maksymalnemu wykorzystaniu pojedynczej krótkiej ekspozycji skóry na promienie UVB do zapoczątkowania reakcji syntezy witaminy D, która w dalszych etapach nie wymaga energii słonecznej.
Przedłużona ekspozycja skóry na promienie UV prowadzi do powstania nieaktywnych biologicznie związków, lumisterolu i tachysterolu, które zapobiegają niebezpieczeństwu zatrucia witaminą D3 na skutek nadmiernego nasłonecznienia. Nieaktywne biologicznie fotoprodukty, wobec braku zdolności DBP do ich wiązania, ze złuszczającym się naskórkiem systematycznie są usuwane z organizmu.
Należy podkreślić, że przemiana pre-D3 do lumisterolu i tachysterolu pod wpływem promieni UV jest odwracalna. W przypadku obniżenia zawartości pre-D3 (w wyniku jej powolnej przemiany termicznej do witaminy D3) jej ilość może wzrosnąć na skutek fotochemicznej przemiany lumisterolu do pre-D3.
W badaniach klinicznych nad efektywnością skórnej syntezy witaminy D wykazano, że u osób o jasnym zabarwieniu skóry 1 dawka rumieniowa (1 MED), tj. dawka promieni UV wywołująca minimalny rumień skóry, doprowadza do 10-krotnego wzrostu stężenia witaminy D3 w surowicy krwi na skutek uwolnienia około 30 μg D3 z 1 m2 powierzchni ciała w ciągu 24 h. Powrót do normalnego stężenia witaminy D3 podwyższonego po naświetleniu skóry promieniami UV następuje po kilku dniach. Zmianom tym u ludzi zdrowych towarzyszy niewielki wzrost poziomu 25(OH)D; u osób z niedoborami witaminy D obserwuje się natomiast znaczny, bo aż 3-krotny, wzrost stężenia 25(OH)D w krwi.
Metabolizm witaminy D
Pierwszym etapem na drodze metabolizmu witaminy D jest jej hydroksylacja w pozycji 25 łańcucha bocznego, doprowadzająca do powstania 25-hydroksywitaminy D – 25(OH)D. Reakcja ta zachodzi w wątrobie – zarówno we frakcji mitochondrialnej, jak i mikrosomalnej hepatocytów, gdzie zlokalizowane są 25-hydroksylazy różniące się składem i powinowactwem z substratem (stałą Micheaelisa – Km).
W fizjologicznym stężeniu 25(OH)D nie ma biologicznego wpływu na metabolizm wapnia. Ten metabolit witaminy D wymaga dalszej hydroksylacji do powstania czynnego związku 1,25-dihydroksywitaminy D-1,25(OH)2D (ryc. 1).
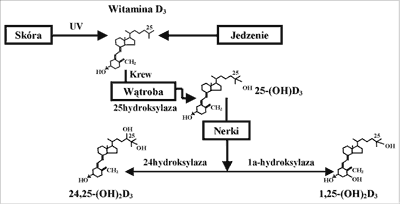
Ryc. 1. Przemiany witaminy D.
Nerka jest głównym miejscem syntezy 1,25(OH)2D. Synteza 1,25(OH)2D w nerkach zależna jest od wielofunkcyjnego receptora klirensującego – megaliny, występującej w cewkach proksymalnych. Megalina cewkowa wiążąc przesączoną w kłębuszkach 25(OH)D (związaną z białkiem nośnikowym) przekazuje ją do komórek cewkowych, gdzie zachodzi konwersja 25(OH)D do 1,25(OH)2D. Megalina nerkowa wiąże również parathormon (PTH), uczestnicząc w jego inaktywacji oraz bierze udział w regulacji aktywności receptora dla PTH i peptydu PTH-podobnego (PTH – PTHrP – R). Ustalono, że w nerce oprócz 1,25(OH)2D mogą powstać również inne dwuhydroksylowe pochodne witaminy D, jak 24, 25-dihydroksywitamina D (24, 25(OH)2D) w wyniku działania 24-hydroksylazy) oraz 25, 26(OH)2D (w wyniku działania 26-hydroksylazy). Pozanerkową konwersję 25(OH)D do 1,25(OH)2D wykazano w tkance kostnej, łożysku, monocytach i tkankach ziarniczych (1, 2, 4).
Mechanizmy regulujące metabolizm witaminy D
W odróżnieniu od nieenzymatycznej syntezy witaminy D w skórze, przemiana witaminy D do 25(OH)D i 1,25(OH)2D wymaga obecności enzymów – hydroksylaz, których aktywność zależy od wielu czynników. Ustalono, że aktywność 25-hydroksylazy wątrobowej ulega zwiększeniu pod wpływem ilości substratu, stężenia DBP i niektórych leków (np. przeciwpadaczkowych), a zmniejszeniu przez końcowe metabolity tej przemiany. Aktywność enzymu katalizującego 1-hydroksylację i/lub 24-hydroksylację 25(OH)D zależy natomiast od stężenia wapnia i fosforanów w surowicy krwi oraz od wielu hormonów i prostaglandyn.
Najaktywniejszym stymulatorem syntezy 1,25(OH)2D jest parathormon (PTH) i peptyd PTH-podobny (PTHrP) oraz obniżenie kalcemii i fosfatemii. Działanie hamujące na powstawanie 1,25(OH)2D ma wzrost stężenia tego hormonu (1,25(OH)2D tworzy z 1α-hydroksylazą układ sprzężenia zwrotnego), niedobór PTH i PTHrP, hiperkalcemia, hiperfosfatemia, kalcytonina, a także kwasicza konstelacja metaboliczna.
Czynniki pobudzające 1α-hydroksylację 25(OH)D wykazują najczęściej hamujący wpływ na powstawanie 24,25(OH)2D i odwrotnie – inhibitory 1α-hydroksylazy 25(OH)D z reguły stymulują syntezę 24,25(OH)2D (1, 4).
Katabolizm metabolitów witaminy D
W warunkach fizjologicznych w surowicy krwi obecne są trzy główne metabolity witaminy D: 25(OH)D, 1,25(OH)2D i 24,25(OH)2D, które w miarę upływu czasu są eliminowane z ustroju. Odbywa się to na skutek konwersji w związki bardziej polarne w tkankach docelowych, a także wydalania z żółcią. Metabolit wątrobowy 25(OH)D ulega eliminacji przez 1- i 24-hydroksylację oraz wydaleniu z żółcią, po uprzednim jego sprzężeniu z kwasem glukuronowym lub siarkowym.
Podobnie jak 25(OH)D, pozostałe metabolity witaminy D po sprzężeniu z kwasem glukuronowym lub siarkowym w wątrobie, są wydalane wraz z żółcią do jelita i podlegają krążeniu jelitowo-wątrobowemu (analogicznie do kwasów żółciowych). Ustalono, że w warunkach fizjologicznych zaledwie 3% z krążących w krwi metabolitów ulega wydaleniu z moczem i z kałem. Tak więc stężenie metabolitów witaminy D w krwi jest determinowane nie tylko przez ich syntezę, ale i przez katabolizm oraz wydalanie (2, 4).
Znaczenie biologiczne 1,25(OH)2D
W ostatnich latach udowodniono jednoznacznie wiodącą rolę metabolitów witaminy D w regulacji gospodarki wapniowo-fosforanowej przez oddziaływanie ich na jelito, nerki, kości, skórę, przytarczyce.
Dzięki odkryciu receptorów specyficznych dla 1,25(OH)2D poznano bliżej mechanizm działania witaminy D w regulacji homeostazy wapniowo-fosforanowej. Jest on podobny do działania większości hormonów steroidowych. Cząsteczka steroidu wnika do komórki receptorowej na zasadzie dyfuzji niejonowej (lipofilna z łatwością pokonuje bariery lipidowe), łączy się z białkiem receptorowym i jako zaktywowany kompleks indukuje w jądrze biosyntezę nowej molekuły mRNA, a następnie nowego białka lub białek. Ten sposób działania ma miejsce we wszystkich komórkach tkanek klasycznie docelowych dla witaminy D: w jelicie, kościach, nerkach. Efekty biologiczne 1,25(OH)2D zależne są od liczby receptorów (VDR) i ich powinowactwa do 1,25(OH)2D, heterodimeryzacji kompleksu VDR – 1,25(OH)2D z receptorem retinoidowym X i powinowactwem tego heterodimeru do odpowiedniego elementu odpowiedzi na poziomie DNA jądrowego oraz od katabolizmu kalcitriolu. Jedną z najważniejszych funkcji 1,25(OH)2D jest wpływ tego hormonu na jelito, nerkę i kość (1, 5, 6).
Wpływ 1,25(OH)2D na jelito
Kalcytriol (1,25(OH)2D) działa na enterocyt w sposób bezpośredni i pośredni, doprowadzając w efekcie do wzrostu absorpcji wapnia z przewodu pokarmowego. Mechanizm bezpośredniego oddziaływania kalcytriolu polega na modyfikacji struktury fosfolipidów błony luminalnej, co może powodować lokalny wzrost przepuszczalności błony luminalnej dla jonów. Ten etap działania witaminy D jest niezależny od syntezy białek de novo.
Drugiemu pośredniemu mechanizmowi działania 1,25(OH)2D na poziomie komórki towarzyszy aktywacja genomu i synteza de novo białka wiążącego wapń CaBP.
Czas biologicznej reakcji jelita w postaci wzrostu absorpcji wapnia na podanie 1,25(OH)2D pojawia się po 8-10 godzinach. W wyniku obserwacji odpowiedzi jelita na różne dawki 1,25(OH)2D podawane w odstępach 3-tygodniowych ustalono, że istnieje zależność liniowa między jelitową akumulacją 1,25(OH)2D a ilością stężenia jelitowego CaBP. Oznacza to, że im więcej 1,25(OH)2D lokalizuje się w jelicie, tym większa jest produkcja CaBP, tym większe jest również wchłanianie wapnia z przewodu pokarmowego.
Udowodniono jeszcze jeden bardzo ważny aspekt działania witaminy D jako systemu endokrynnego, a mianowicie: wpływ zawartości wapnia i fosforu w diecie na stężenie jelitowe CaBP, a tym samym na absorpcję wapnia z przewodu pokarmowego (AbCa).
Wykazano, że jelito odznacza się dużymi zdolnościami adaptacyjnymi w tym zakresie. Przy stosowaniu diety o niskiej zawartości wapnia jelito wytwarza znacznie więcej CaBP niż przy podawaniu diet o dużej zawartości wapnia, gdy synteza CaBP jest kilka razy mniejsza. W przypadku stosowania diety o niskiej zawartości wapnia dochodzi do hipokalcemii, która jest bodźcem do wydzielania parathormonu (PTH) przez przytarczyce. Parathormon natomiast jest silnym stymulatorem działania 1α-hydroksylazy nerkowej, doprowadzającym do wzrostu syntezy 1,25(OH)2D. Z chwilą normalizacji stężenia wapnia w surowicy zmniejsza się synteza PTH i dzięki temu zmniejsza się aktywność 1α-hydroksylazy nerkowej, doprowadzając do zmniejszonej syntezy 1,25(OH)2D, spadku syntezy CaBP w jelicie, co w ostateczności prowadzi do spadku absorpcji wapnia w przewodzie pokarmowym (1, 4).
Wpływ 1,25(OH)2D na nerkę
Nerki są miejscem biosyntezy 1,25(OH)2D oraz organem jej docelowego działania. 1,25(OH)2D stymuluje wchłanianie zwrotne wapnia i fosforanów z płynu cewkowego do krwi. Jak już wspomniano, 1,25(OH)2D wpływa pobudzająco na aktywność 24-hydroksylazy, a hamująco na 1α-hydroksylazę (2, 4).
Wpływ 1,25(OH)2D na tkankę kostną
Powszechnie wiadomo, że witamina D niezbędna jest do prawidłowego rozwoju i mineralizacji kości. Receptory dla 1,25(OH)2D oraz białko wiążące wapń (CaBP) o ciężarze cząsteczkowym 28 tys. daltonów odkryto w osteoblastach i chondrocytach. 1,25(OH)2D stymuluje receptory jądrowe witaminy D w osteoblastach, zwiększając ekspresję ligandu aktywującego jądrowy czynnik transkrypcyjny κB (NFκB) RANKL. Stwierdzono ich obecność również w prekursorach osteoklastów, w monocytach, ale nie znaleziono w dojrzałych osteoklastach, co sugeruje, że 1,25(OH)2D wywiera wpływ na formowanie osteoklastów, nie wpływa natomiast na ich aktywność.
Niedobór tego hormonu jest przyczyną krzywicy u dzieci, a u osób dorosłych osteomalacji lub osteoporozy. Nadmiar 1,25(OH)2D powoduje wzrost resorpcji kostnej (osteolizę osteoklastyczną) i hamuje syntezę kolagenu, naśladując w swym działaniu efekt jaki PTH wywiera na kość; nadmiar 1,25(OH)2D poza tym może blokować replikację komórek i hamować wzrastanie kości (2, 4).
NIEDOBORY WITAMINY D
Przyczyny niedoboru witaminy D
Niedobór witaminy D jest następstwem: zmniejszonego jej „dowozu” z pożywieniem, upośledzonej syntezy skórnej, zaburzonej hydroksylacji, nadmiernego katabolizmu. Może się ujawnić (mimo dostarczania witaminy D w dostatecznej ilości) na skutek zmniejszonej wrażliwości tkanek docelowych na 1,25(OH)2D (np. defekt receptora). Prowadzi on do hipokalcemii, rozwoju wtórnej nadczynności przytarczyc i hipofosfatemii, zaburzeń dojrzewania i mineralizacji nowo powstającej tkanki kostnej w miejscach jej przebudowy bądź gromadzenia się tkanki chrzęstnej w kościach dzieci rosnących.
Krzywica z niedoboru witaminy D u dzieci, a osteomalacja u dorosłych, często jeszcze występują wśród osób o ciemnym zabarwieniu skóry, żyjących w krajach o umiarkowanym klimacie oraz u osób starszych, stale przebywających w zamkniętych pomieszczeniach. Przyczyną niedoborów witaminy D u tych osób jest niedostateczna synteza skórna witaminy D i/lub niedobory egzogennej witaminy D podawanej z pożywieniem.
Do niedoboru witaminy D dochodzi u osób z zespołem złego wchłaniania, który zazwyczaj ujawnia się u osób z przewlekłymi chorobami przewodu pokarmowego lub przewlekłymi chorobami wątroby. Ustalono, że w tej grupie dominują zaburzenia metabolizmu witaminy D, spowodowane jej zmniejszoną biodostępnością w wyniku upośledzonej syntezy skórnej, upośledzonego wchłaniania z przewodu pokarmowego, zaburzeń w krążeniu jelitowo-wątrobowym witaminy D i jej metabolitów. Niskie stężenie 25(OH)D u osób z marskością wątroby, które ulega zwiększeniu po podaniu witaminy D drogą pozajelitową sugeruje, że w większości przypadków w tej grupie chorych przyczyną niedoborów witaminy D jest jej upośledzona biodostępność, a nie zaburzona hydroksylacja wątrobowa. Do zaburzeń hydroksylacji witaminy D do 25(OH)D w wątrobie dochodzi bowiem tylko w skrajnie ciężkiej niewyrównanej marskości wątroby.
Krzywica lub osteomalacja po lekach przeciwdrgawkowych stanowi, obok ciężkich chorób wątroby, jeden z nielicznych przykładów zaburzenia metabolizmu witaminy D3 na etapie jej wątrobowej hydroksylacji. Ustalono, że barbiturany, fenydantoina i piramidon są induktorami mikrosomalnych enzymów wątrobowych (np. glukuronidaz), do których należy również nadzorująca wątrobowy etap aktywacji witaminy D 25-hydroksylaza cholekalcyferolu. Indukcja wątrobowej hydroksylacji witaminy D3 oddziałuje niekorzystnie, przyspiesza bowiem wydalanie z żółcią glukuronowych nieaktywnych metabolitów witaminy D. U pacjentów przewlekle leczonych przeciwdrgawkowo stwierdzono niskie stężenie 25(OH)D w surowicy i zmieniony jej stosunek do 1,25(OH)2D, ale stężenie 1,25(OH)2D jest prawidłowe lub wysokie.
Zaburzenia metabolizmu witaminy D na etapie jej 1α-hydroksylacji prowadzą do znacznego obniżenia stężenia 1,25(OH)2D i ujawnienia objawów niedoboru witaminy D. Występują one zarówno w chorobach dziedzicznych – witamino-D zależnej krzywicy typu II, w krzywicy hipofosfatemicznej, jak i w nabytych – w przewlekłej niewydolności nerek, w ciężkich tubulopatiach, w niedoczynności przytarczyc, w rzekomej niedoczynności przytarczyc. Obniżona aktywność 1α-hydroksylazy jest przyczyną zmniejszonego stężenia 1,25(OH)2D w osteoporozie, zwłaszcza starczej, zaburzeniach wydzielania GH, prolaktyny oraz insuliny (1, 2, 4).
Najbardziej reprezentatywnym zespołem chorobowym, w którym dochodzi do zaburzeń receptora tkankowego 1,25(OH)2D, jest krzywica witamino-D zależna typu II. Zespół ten jest uwarunkowany genetycznie. Cechuje go obniżona absorpcja wapnia z przewodu pokarmowego – mimo bardzo wysokiego stężenia 1,25(OH)2D w krwi. Przyczyn nieprawidłowej odpowiedzi jelita na 1,25(OH)2D upatruje się w zmniejszeniu liczby receptorów dla 1,25(OH)2D w nabłonku jelitowym i/lub w zaburzeniach – w połączeniu 1,25(OH)2D z białkiem receptorowym albo w zaburzeniach w wewnątrzkomórkowym przesuwaniu kompleksu 1,25(OH)2D z receptorem. Zaburzenia te nie zapewniają syntezy dostatecznej ilości CaBP – niezbędnego do wchłonięcia wapnia z przewodu pokarmowego, prowadząc do hipokalcemii, hipersekrecji parathormonu, który z kolei wzmaga fosfaturię i mobilizację mineralnych składników kości. W rezultacie rozwija się pełen obraz klinicznego niedoboru witaminy D, mimo jej dostatecznej podaży, czego wykładnikiem jest wysokie stężenie w surowicy krwi zarówno 25(OH)D, jak i 1,25(OH)2D (2, 7).
Wskaźniki biochemiczne niedoboru witaminy D i skutki jej niedoboru dla kości
Oznaczenie stężenia 25-hydroksywitaminy D w surowicy krwi jest najlepszym, biochemicznym wskaźnikiem odzwierciedlającym zasoby witaminy D w organizmie. Oznaczanie stężenia najbardziej aktywnego biologicznie metabolitu witaminy D tj. 1,25(OH)2D jest nieprzydatne, a nawet zwodnicze, ponieważ przy niedoborze witaminy D może być ono prawidłowe a nawet podwyższone. Nie bez znaczenia jest także fakt, że: jego stężenie jest 1000 razy mniejsze niż 25(OH)D, ma krótki czas półtrwania (4-6 godz. vs 2 tyg.).
Należy podkreślić, że ustalone i podawane przez referencyjne laboratoria, jako prawidłowe, stężenia witaminy D (10-55 ng/ml) w świetle obecnej wiedzy są nieprawidłowe. Dowiedziono bowiem, że pomimo stężenia 25(OH)D w granicach normy laboratoryjnej, wiele osób dorosłych ma ogólnoustrojowy niedobór witaminy D, który prowadzi do wtórnej nadczynności przytarczyc z następową, stałą, wzmożoną resorpcją kości .
Obserwacje, że wydzielanie PTH jest minimalne, a efektywność wchłaniania wapnia w jelitach największa przy stężeniu 25(OH)D przekraczającym 30 ng/ml (75 nmol/l), stała się podstawą uznania za minimalne prawidłowe stężenie 25(OH)D – wartość 30 ng/ml (75 nmol/l). Wartości 21-29 ng/ml uważa się za niewystarczające, ponieważ nie powodują odpowiedniej supresji PTH, ani nie wiążą się z optymalnym wchłanianiem wapnia w jelitach. Stężenie 25(OH)D poniżej 20 ng/ml (50 nmol/l) uważane jest za granicę niedoboru witaminy D (8, 9).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Marcinowska-Suchowierska E: Witamina D – aktualny stan wiedzy. Wykorzystanie witaminy D profilaktyce i leczeniu osteoporozy. Polskie Archiwum Medycyny Wewnętrznej. 2002; CVII, 2 (2): 111-119.
2. Bouillon R: Vitamin D metabolism and action. Osteoporosis Int 1998; 8, 13.
3. Marcinowska-Suchowierska E: Skuteczność skórnej syntezy witaminy D u osób z przewlekłymi chorobami przewodu pokarmowego. Praca Habilitacyjna. CMKP 1989.
4. Brown AJ: Regulation of vitamin D action (editorial). Nephrol Dial Transplant 1999; 14, 11.
5. Hilpert J, et al.: Megalin antagonizes activation of the parathyroid hormone receptor. J. Biol. Chem., 1999; 274, S620.
6. Matsuyama T: Vitamin D receptor genotypes and bone mineral density. Lancet 1995; 345, 1238.
7. Riggs BL: Effect of long term treatment with calcitriol on calcium absorption and mineral metabolism in postmenopausal osteoporosis. J. Clin. Endocrinol. Metab., 1985; 61, 457.
8. Holic MF: High Prevalence of Vitamin D Inadequacy and Implications for Health; Mayo Clin. Proc., 2006; 81: 353-373.
9. Marcinowska-Suchowierska E: komentarz do artykułu: Rola Witaminy D w utrzymaniu zdrowych kości oraz w profilaktyce złamań. Medycyna po Dyplomie 2007; 16: 127-129.
10. Vieth R: Problems with direct 25(OH)D assays, and three target amount vitamin D nutrition desirable for patients with osteoporosis. Osteoporosis Int. 2000; 11, 635.
11. Vieth R: What is the optimal vitamin D status for health? Progress in Biophysics and Molecular Biology 2006; 92: 26-32.
12. MacFarlane GD, et al.: Hypovitaminosis D in a normal, apparently healthy urban European population. Journal of Steroid Biochemistry & Molecular Biology 2004; 89-90: 621-622.
13. Webb AR, et al.: Influence of season and latitude on the cutaneous synthesis of vitamin D3: exposure to winter sunlight in Boston and Edmonton will not promote vitamin D3 synthesis in human skin. J. Clin. Endocrinol. Metab., 1988; 67: 373-378.
14. Matsuoka LY, et al.: Sunscreens suppress cutaneous vitamin D3 synthesis; J. Clin. Endocrinol. Metab., Jun 1987; 64: 1165-1168.
15. Gloth FM, et al: Vitamin D deficiency in homebound elderly persons; JAMA, Dec., 1995; 274: 1683-1686.
16. Lamberg-Allardt Ch: Vitamin D in foods and as supplements; Biophysics and Molecular Biology 2006; 92: 33-38.
17. Arunabh S, et al.: Body Fat Content and 25-Hydroxyvitamin D Levels in Healthy Women; J. Clin. Endocrinol. Metab., 2003; 88(1):157-161.
18. Czerwińska E, et al.: Calcium homeostasis and biochemical markers of bone turnover In patients with morbid obesity. Obesity Reviews 2006; 7 (Suppl 2): 139.
19. Wortsman J, et al: Decreased bioavailability of vitamin D in obesity. Am. J. Clin. Nutr., 2000; 72: 690-693.
20. Bell NH, et al.: Evidence for alteration of the vitamin D-endocrine system in obese subjects. J. Clin. Invest., 1985; 76: 370-373.
21. Malinowski SS: Nutritional and metabolic complications of bariatric surgery Am. J. Med. Sci., 2006 Apr; 331(4): 219-25.
22. Holik MF: Resurrection of vitamin D deficiency and rickets; J. Clin. Invest., 2006; 116: 2062-2072.
23. Bruyere O, et al: Prevalence of vitamin D inadequacy is high in European postmenopausal women. Osteoporosis Int 2007; 18 (Suppl. 1): S29-S175 P297.
24. Lips P, et al.: The prevalence of vitamin D inadequacy amongst women with osteoporosis: an international epidemiological investigation Journal of Internal Medicine 2006; 260: 245-254.
25. Compston J (European Union Osteoporosis Consultation Panel): Action Plan for the prevention of osteoporotic fractures in the European Community; Osteoporos Int 2004; 15: 259-262.
