© Borgis - Anestezjologia Intensywna Terapia 3/2001, s. 181-190
Ewa Karpel
Mediatory ogólnoustrojowej odpowiedzi zapalnej – znaczenie w praktyce klinicznej intensywnej terapii
The place of systemic inflammatory response mediators in intensive care
z Katedry i Klinika Anestezjologii i Intensywnej Terapii;
kierownik: prof. dr hab. A. Dyaczyńska-Herman – Śl AM w Katowicach

Zespół ogólnoustrojowej reakcji zapalnej (SIRS – systemic inflammatory response syndrome) rozpoznawany jest u 50 – 70% chorych leczonych w oddziałach intensywnej terapii i aktualnie stanowi największy problem diagnostyczno-terapeutyczny w praktyce klinicznej. Wielorakość przyczyn odpowiedzialnych za jego występowanie oraz zróżnicowany przebieg kliniczny i wyniki leczenia inspirują podejmowanie badań w celu poznania złożonych mechanizmów zaangażowanych w patomechanizm SIRS [1,2,3,4,5].
Reakcja zapalna
Zapalenie jest jednym z najstarszych pojęć w patofizjologii chorób i cierpienia. Definiuje się je jako odpowiedź na czynnik zewnętrzny ingerujący w naturalną homeostazę ustroju. W ostatnim dwudziestoleciu poznano wiele mechanizmów tego złożonego procesu. Chociaż reakcja zapalna jest jednym z mechanizmów obronnych ustroju przed szkodliwymi czynnikami, jest związana z nieprzyjemnymi odczuciami chorego, często powoduje cierpienie, a zdarza się, że nie prowadzi do wyzdrowienia – wręcz przeciwnie do stanu ciężkich, zagrażających życiu zaburzeń homeostazy. Ból, obrzęk, przekrwienie, gorączka i upośledzenie czynności czasem nie tylko nie ustępują w miarę procesu leczenia, lecz obejmują coraz to nowe okolice ciała prowadząc do nasilenia objawów chorobowych. Ustrój przegrywa walkę z chorobą [6].
Kluczową rolę w reakcji zapalnej odgrywa układ immunologiczny, którego anatomia i fizjologia dostosowane są do pełnienia funkcji obronnej przed działaniem szkodliwych dla organizmu czynników. Narządy układu immunologicznego: szpik, grasica, migdałki, wątroba, śledziona i rozsiane w całym organizmie grudki chłonne produkują komórki immunologiczne, które drogą krwi docierają wszędzie tam, gdzie wystąpi potrzeba obrony przed szkodliwym dla ustroju czynnikiem. Monocyty/makrofagi, limfocyty B, T, NK i cytoktoksyczne oraz granulocyty stanowią podstawowe elementy komórkowej odpowiedzi immunologicznej na czynnik zapalny [6].
Fundamentalną rolę w funkcji obronnej odgrywają chemiczne czynniki humoralne stanowiące system wczesnego ostrzegania o niebezpieczeństwie i jednocześnie uczestniczące w pierwotnych mechanizmach walki z czynnikiem uszkadzającym.
Funkcjonalnie dzieli się je zależnie od mechanizmu aktywacji na następujące grupy:
– osoczowe układy krzepnięcia, kinin i dopełniacza aktywowane przez czynnik zapalny, które wpływają na proces gojenia się ran, tamowania krwotoku, odpowiedniej gry naczyniowej i uczynnienia białek ostrej fazy,
– wewnątrzkomórkowe biochemiczne substancje chemiczne (histamina, serotonina, enzymy lizosomalne), które pod wpływem czynnika zapalnego zostają uwalniane z komórek wywołując specyficzne zmiany metabolizmu komórkowego i przepływu krwi w miejscu objętym procesem zapalnym,
– produkowane przez pobudzone czynnikiem zapalnym komórki układu immunologicznego, a wśród nich głównie monocyty/makrofagi, specyficzne zapalne proteiny, nie syntetyzowane w stanie zdrowia, których funkcja polega na modulacji różnych systemów obronnych oraz na przekazywaniu informacji o zadziałaniu czynnika uszkadzającego innym komórkom i narządom organizmu. Zalicza się do nich metabolity kwasu arachidonowego (prostaglandyny, leukotrieny), czynnik aktywujący płytki PAF ( platelet activating factor) i cytokiny.
Przebieg ostrej miejscowej reakcji zapalnej obejmuje:
– zmiany w przepływie krwi przez tkankę objętą zapaleniem, spowodowane grą naczyniową polegającą na krótkotrwałym kurczu arterioli, a następnie ich rozszerzeniu odpowiedzialnym za przekrwienie objawiające się klinicznie zaczerwienieniem,
– zmiany w przepuszczalności ścian naczyniowych dla białek powodujące spadek ciśnienia osmotycznego krwi, wychodzenie płynu do przestrzeni pozanaczyniowej i obrzęk tkanek, marginację leukocytów, adhezję do śródbłonka naczyniowego limfocytów, monocytów i granulocytów, migrację komórek immunologicznych w kierunku zwiększonego stężenia czynnika chemotaktycznego.
Przebieg reakcji zapalnej jest sterowany przez system mediatorów stymulujących układ granulocytów i monocytów/makrofagów powodując ich wędrówkę do źródła zapalenia (chemotaksja) oraz nadając im właściwości adhezyjne i opsonizujące białka antygenów bakteryjnych. Pobudzony równocześnie układ krzepnięcia odgrywa istotną rolę w miejscowym procesie gojenia ran. Charakterystyczna dla procesu zapalenia gra naczyniowa, mająca na celu miejscowe przekrwienie i zwiększenie przepuszczalności naczyń jest również efektem działania kompleksu mediatorów humoralnych. Najsilniejszym i najlepiej poznanym mechanizmem aktywacji ogólnoustrojowej odpowiedzi zapalnej jest stymulacja zapalenia przez zakażenie, wywoływana przez działanie lipopolisacharydowych cząsteczek endotoksyn bakteryjnych (ryc. 1) [6,7,8,9].
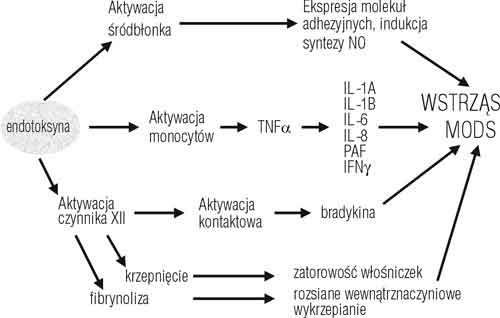
Ryc. 1. Kaskada mediatorów ogólnoustrojowej reakcji zapalnej
Dla zmian zapalnych o charakterze ogólnoustrojowym najistotniejsza jest ta funkcja mediatorów, która polega na przekazywaniu informacji o zadziałaniu czynnika zapalnego innym komórkom i tkankom. Funkcja ta leży u podstaw patofizjologii ciężkich ogólnoustrojowych reakcji zapalnych. Gdy czynnik wywołujący zapalenie jest bardzo silny, działa przez dłuższy czas i dotyczy pierwotnie narządów o istotnym znaczeniu fizjologicznym oraz rozbudowanym unaczynieniu, istnieje zagrożenie stymulacji ogólnoustrojowej odpowiedzi zapalnej obejmującej wszystkie funkcje układowe.
Tak długo, jak długo czynnik zapalny działa miejscowo, aktywność pobudzonych mediatorów jest korzystna dla procesu zdrowienia i gojenia się ran, a jego efektem jest najczęściej wyzdrowienie. Gdy czynnik zapalny zadziała ogólnie (np. zakażenie krwi) lub jego działanie miejscowe jest bardzo silne i długotrwałe, mediatory zapalenia powodują objęcie procesem zapalnym innych, nie narażonych bezpośrednio na szkodliwe działanie czynnika zapalnego tkanek i narządów, a przebieg choroby staje się uogólniony, cięższy, długotrwały i o znacznie gorszym rokowaniu (tab. I.).
Tab. I Miejscowe i ogólnoustrojowe efekty działania mediatorów zapalnych
| Działanie | W miejscu zadziałania | Ogólne |
| Objawy zapalne | Miejscowe objawy zapalne | Ogólne objawy zapalne |
| Reakcja naczyniowa | Przekrwienie | Spadek systemowego oporu naczyniowego |
| Przepuszczalność naczyń | Obrzęk miejscowy | Utrata wody wewnątrznaczyniowej - spadek objętości krwi |
| Układ krzepnięcia | Zatrzymanie krwawienia | Wykrzepianie wewnątrznaczyniowe |
| Aktywacja mediatorów | w miejscu zapalenia | w krążeniu systemowym |
| Efekt reakcji zapalnej | Zdrowienie | SIRS, czasem MODS |
Kluczową rolę w uogólnianiu procesu zapalnego odgrywa śródbłonek naczyń krwionośnych (ostatnio przez niektórych autorów zwany narządem), którego aktywacja przez mediatory zapalenia jest już udokumentowana, a którego aktywność w połączeniu z aktywacją układu krzepnięcia prowadzi do uogólnionych zaburzeń przepływu tkankowego i narządowego, ze wszystkimi jego następstwami [10,11,12,13,14,15].
Definicje ACCP/SCCM
Intensywne badania patofizjologii, objawów klinicznych i możliwości leczenia zespołu ogólnoustrojowej reakcji zapalnej o różnorodnym przebiegu wskazały na potrzebę wprowadzenia do nazewnictwa medycznego nowych terminów opisujących złożoną problematykę tych stanów. W 1991 roku na konferencji uzgodnień zorganizowanej przez Amerykańskie Towarzystwo Pneumonologiczne oraz Towarzystwo Medycyny Stanów Krytycznych ( American College of Chest Physicians i Society for Critical Care Medicine) ustalono międzynarodowe nazewnictwo dotyczące posocznicy.
Uzgodniono i przyjęto następujące definicje [16]:
1) zespół ogólnoustrojowej reakcji zapalnej (SIRS) – jest ogólnoustrojową odpowiedzią o charakterze zapalnym spowodowaną różnorodnymi czynnikami i charakteryzującą się występowaniem co najmniej dwóch z następujących objawów: ciepłota>38°C (lub<36°C), częstość akcji serca>90/min., częstość oddychania> 20/min. (lub PaCO2<32 mmHg; 4,3 kPa), liczba leukocytów> 12 000/mm3 (lub <4 000/mm3) lub> 10% niedojrzałych granulocytów obojętnochłonnych. SIRS rozpoznaje się u chorego spełniającego dwa z powyższych kryteriów niezależnie od przyczyny je wywołującej;
2) posocznica ( sepsis) – jest ogólnoustrojową odpowiedzią zapalną na czynnik mikrobiologiczny. Jest to SIRS wywołany zakażeniem potwierdzonym izolacją patogennych organizmów z miejsc, w których nie powinny bytować;
3) posocznica o ciężkim przebiegu ( severe sepsis) – objawia się spadkiem ciśnienia tętniczego, hipoperfuzją i dysfunkcją narządową. Spadek ciśnienia rozpoznaje się, gdy ciśnienie skurczowe wynosi <90 mmHg (12 kPa) lub stwierdza się obniżenie ciśnienia skurczowego o więcej niż 40 mmHg (5,3 kPa) w stosunku do wartości wyjściowej. Hipoperfuzja charakteryzuje się podwyższonym poziomem mleczanów, skąpomoczem lub zaburzeniami świadomości;
4) wstrząs septyczny (septic shock) – charakteryzuje się spadkiem ciśnienia w przebiegu posocznicy utrzymującym się pomimo adekwatnej terapii płynami wypełniającymi łożysko naczyniowe i wywołującym zaburzenia perfuzji lub dysfunkcję narządową. Pacjenci, którzy po zastosowaniu leków wazoaktywnych nie wykazują spadku ciśnienia tętniczego, ale demonstrują objawy hipoperfuzji i dysfunkcji narządowej są również traktowani jako chorzy we wstrząsie septycznym [16].
Przyjęte nazewnictwo zalicza do szeroko rozumianego stanu ogólnoustrojowej odpowiedzi zapalnej wszystkie, o różnym stopniu nasilenia zespoły chorobowe związane z ciężkim zakażeniem, chociaż nie stanowią one wyłącznych czynników wywołujących SIRS. W przebieg reakcji, niezależnie od przyczyny wyjściowej zaangażowane są te same produkowane i uwalniane do krwiobiegu mediatory, a ich ogólnoustrojowe efekty uszkadzające funkcje narządów często prowadzą do zespołu zaburzeń o bardzo zbliżonym przebiegu. Tylko charakter pierwotnego czynnika uszkadzającego różnicuje te stany. Uraz (w tym operacyjny), ostre niedokrwienie, oparzenia, zatrucie, ostre zapalenie trzustki, reakcje odrzucenia przeszczepu mogą uruchamiać tę samą co zakażenie kaskadę mediatorów humoralnych i komórkowych, a przez to prowadzić do obrazu klinicznego postępujących zaburzeń o przebiegu zbliżonym w ocenie klinicznej do zawartej w definicjach ACCP/SCCM sekwencji rozwoju zaburzeń ogólnoustrojowych prowadzących w efekcie do zespołu niewydolności wielonarządowej (MODS – multiorgan dysfunction syndrome) [19,21,30, 39] (ryc. 2).
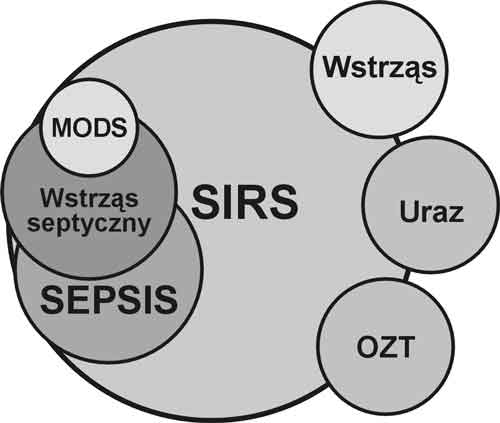
Ryc. 2. Ogólnoustrojowa reakcja zapalna i stopnie jej zaawansowania – wg. ACCP/SCCM
Zespół zaburzeń funkcji wielu narządów jest najcięższym stanem klinicznym ogólnoustrojowej odpowiedzi zapalnej, wymagającym zastosowania wszystkich możliwych metod intensywnej terapii i mimo ogromnego postępu w tej dziedzinie obarczonym ciągle jeszcze dużą śmiertelnością. Przyjęta definicja na konferencji uzgodnień ACCP/SCCM MODS dotyczy stanu tak zaawansowanych zaburzeń funkcji narządowych u krytycznie chorego, w którym bez intensywnej interwencji terapeutycznej homeostaza nie może zostać przywrócona.
Mediatory reakcji zapalnej
Mechanizm zapłonowy ogólnoustrojowej reakcji zapalnej stanowią tak zwane pierwotne cytokiny prozapalne: TNF-alfa ( tumor necrosis factor-alfa) i interleukina-1, które są produktem zaktywowanych przez silny czynnik uszkadzający monocytów/ makrofagów. Najsilniejszym czynnikiem uruchamiającym ich produkcję jest zakażenie, a konkretnie cząsteczki endotoksyn bakteryjnych, które za pośrednictwem białek wiążących lipopolisacharydy (LPS binding protein) wiążą się z odpowiednimi receptorami komórek immunologicznych stymulując je do produkcji mediatorów pierwotnych zapalenia (ryc. 3). Wyprodukowane w dużej ilości mediatory pierwotne stanowią alegoryczny zapalnik wybuchu kaskady reakcji zapalnej z pobudzeniem układu dopełniacza, krzepnięcia, kinin, peptydów wazoaktywnych, metabolitów kwasu arachidonowego oraz innych cytokin. W przypadkach przebiegających z dużą nadprodukcją mediatorów drogą naczyń krwionośnych ta „bomba zapalna” dociera do wszystkich narządów i tkanek inicjując w nich zmiany analogiczne do tych, jakie wystąpiłyby w nich wskutek zakażenia miejscowego.

Ryc. 3. Mechanizm zapłonowy ogólnoustrojowej reakcji zapalnej
Reakcja ta raz zapoczątkowana przebiega lawinowo, prowadząc w efekcie końcowym do uruchomienia wszystkich zapalnych czynników miejscowych. Zaburzenia przepływu obwodowego krwi, rozsiane zmiany zatorowo-zakrzepowe, specyficzne efekty działania tlenku azotu, leukotrienów, elastyny i katepsyny odpowiedzialne są za zmiany narządowe upośledzające ich wydolność.
Obraz kliniczny spowodowany działaniem tej zaktywowanej sieci czynników jest dobrze znany w praktyce oddziałów intensywnej terapii jako zespół niewydolności wielonarządowej (MODS) z ostrą niewydolnością oddechową (ARDS), ostrą niewydolnością nerek (ONN) i wykrzepianiem wewnątrznaczyniowym (DIC) [8,13,16,17,18].
Cytokiny
Wśród mediatorów reakcji zapalnej kluczową rolę odgrywają cytokiny – białka regulujące różne procesy biologiczne i ogrywające istotną rolę w patogenezie wielu chorób. Badania ostatnich lat ogniskowały się na ich izolowaniu, poznawaniu mechanizmów stymulujących, efektów działania, wzajemnych powiązań i ewentualnych metod modulowania ich aktywności.
Cytokiny są to hormonopodobne peptydy, produkowane zasadniczo przez immunologiczne komórki zaktywowane czynnikiem zapalnym; w odróżnieniu od endokrynnych hormonów są parakrynami, czyli stanowią nośnik informacji przekazywanej innej komórce nie drogą krwi, a drogą bezpośredniego sąsiedztwa. W przypadku długotrwałego działania silnego bodźca zapalnego produkcja cytokin jest nadmierna, dostają się do osocza i krążąc z krwią wywierają działanie endokrynne. Stanowią wtedy nośnik informacji o zadziałaniu czynnika zapalnego w którymś miejscu ustroju; przekazane drogą krwi innym narządom powodują ich paradoksalną, analogiczną do zapalnej odpowiedź.
Cytokiny działają receptorowo; receptory cytokinowe znajdują się we wszystkich komórkach ustroju, a poza tym występują w postaci wolnej we krwi. Tylko związanie cząsteczki cytokiny z receptorem komórkowym daje efekt pobudzenia. Rozpuszczalne receptory wiążąc się z cząsteczkami cytokin neutralizują je. Budowa strukturalna receptorów cytokinowych jest często podobna – wyjaśnia to wielokierunkowe, plejotropowe ich działanie [10,11,18] (ryc. 4).
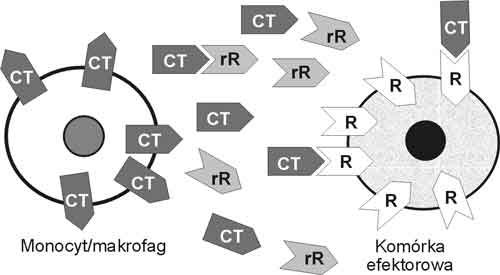
Ryc. 4. Receptorowe działanie cytokin. (CT-cytokina, rR-rozpuszczalny receptor cytokinowy, R-receptor cytokinowy)
Cytokiny wpływają na wszystkie fazy odpowiedzi immunologicznej regulując proliferację, różnicowanie i aktywację limfocytów B, T, komórek NK, monocytów/makrofagów i granulocytów. Regulują one zarówno odpowiedź komórkową jak i humoralną. Wpływają na funkcję dojrzałych neutrofili poprzez aktywację molekuł adhezyjnych i oddziaływanie na migrację tych komórek. Różne cytokiny mogą działać antagonistycznie, addycyjnie lub synergistycznie na te same procesy biologiczne. Dotyczy to zwłaszcza ich wpływu na hematopoezę i reakcję ostrej fazy [12].
Efekty działania cytokin stanowią podstawę opracowania funkcjonalnego podziału ich na grupy protein o zbliżonym, chociaż nie takim samym efekcie.
– mediatory reakcji odpornościowych: TNF-alfa, IL-1, IL-6, IL-8, IFN typu I – stymulują komórki immunologiczne do reakcji zapalnej i produkcji wtórnych mediatorów, przekazują informacje o zadziałaniu bodźca zapalnego sąsiadującym komórkom;
– cytokiny prozapalne: TNF-alfa, TNF-beta, IL-1, IL-8, IL-12, IFN typu II – stymulują odpowiedź zapalną komórkową i humoralną, proliferację limfocytów, produkcję przeciwciał przez komórki B, aktywację, adhezję, agregację neutrofilów, produkcję białek ostrej fazy w wątrobie, podwzgórzową reakcję gorączkową;
– cytokiny przeciwzapalne: IL-4, IL-10, IL-13, TNF-beta – wykazują działanie przeciwzapalne przez hamowanie produkcji mediatorów prozapalnych i stymulację produkcji rozpuszczalnych receptorów cytokin;
– cytokiny regulujące wzrost: IL-2, IL-4, IL-5, IL-12, PDGF, FGF, EDF, TGF – stymulują proliferację i wzrost komórek układu immunologicznego;
– cytokiny aktywujące hematopoezę szpikową: CSFs, IL-6 – stymulują komórki macierzyste szpiku.
Należy podkreślić, że peptydy te działają również wzajemnie na siebie hamując lub stymulując inne cytokiny, a ich złożone sprzężenie wzajemnych relacji jest źródłem przyjętego dla opisania procesu aktywacji i przebiegu odpowiedzi zapalnej terminu „sieć cytokin” [10,19,29,21,22,23].
Przebieg reakcji zapalnej
Reakcja zapalna przebiega w dwóch fazach, które jednakże nie są ściśle rozłożone w czasie:
1. Faza prozapalna: pierwotnym mediatorem jest w tej fazie TNF-alfa, który jest uwalniany we wczesnym okresie jako klucz do odpowiedzi zapalnej, a jego poziom w surowicy koreluje z śmiertelnością w posocznicy. Wykazano, że TNF podany ochotnikom powoduje objawy posocznicy, a przeciwciała przeciw TNF chronią zwierzęta doświadczalne przed objawami wstrząsu po podaniu endotoksyn. Badania kliniczne u ludzi nie potwierdziły tego wyniku przypuszczalnie z powodu zbyt późnego podania przeciwciał.
Do mediatorów prozapalnych zalicza się ponadto cytokiny: IL-1beta, Il-2, IL-6, IL-8, IL-15, IFN-gamma.
2. Faza przeciwzapalna: wzmożona aktywność cytokin prozapalnych stymuluje syntezę cytokin przeciwzapalnych, z których najważniejsze to rozpuszczalne receptory TNF, antagoniści receptorów interleukiny 1 oraz IL-4, IL-10, IL-13, TNF-beta.
Aktywność tych mediatorów koreluje ze śmiertelnością i niewydolnością narządową. Mediatory przeciwzapalne mogą być odpowiedzialne za osłabienie mechanizmu odpowiedzi komórkowej z zaburzoną produkcją cytokin prozapalnych.. Ta niewydolność produkcji cytokin nazywana „immunoparalysis” jest związana ze wzrostem śmiertelności chorych [21,22,23].
W praktyce klinicznej trudno jest te fazy odróżnić, wbrew pozorom zmniejszenie aktywności cytokin prozapalnych na rzecz przeciwzapalnych nie jest korzystne dla chorych i może powodować stan immunosupresji sprzyjający zakażeniom szpitalnym pogarszającym rokowanie.
Próby definiowania stanu reakcji zapalnej polegają na:
– monitorowaniu stopnia ekspresji powierzchniowych molekuł HLA-DR na monocytach,
– ocenie aktywności cytokin pro- i przeciwzapalnych w surowicy,
– badaniu poziomu innych, nie tak specyficznych markerów zapalenia jak: białko C-reaktywne, prokalcytonina, neopteryna, elastaza [11].
Przebieg ogólnoustrojowej odpowiedzi zapalnej jest inicjowany przez aktywację za pośrednictwem receptorów CD14 i TLR ( toll-like receptor) komórek układu immunologicznego: monocytów/makrofagów oraz granulocytów, które:
– produkują i uwalniają cytokiny prozapalne (TNF, IL-1, IL-6, IL-8),
– produkują wolne rodniki tlenowe (nadtlenki), proteazy (elastaza, kolagenaza),
– wydzielają mediatory lipidowe (prostaglandyny, leukotrieny, PAF),
– monocyty wykazujące ekspresję białka powierzchniowego – czynnika tkankowego, który łączy się z czynnikiem VII i inicjuje proces krzepnięcia,
– granulocyty wykazujące ekspresję powierzchniowych cząsteczek adhezyjnych [7,12,23,24,25].
Wśród substancji mediatorowych odpowiedzialnych za przebieg procesu zapalenia można wyróżnić następujące grupy:
– substancje zapłonowe (endotoksyna i inne toksyny) rozpoczynające eksplozję mediatorów i określane jako inicjatory,
– wczesne mediatory (cytokiny: TNF-alfa, IL-1, IL-6, IL-8), produkowane przez granulocyty obojętnochłonne, komórki śródbłonka, monocyty, makrofagi i fibroblasty określane jako aktywatory,
– mediatory wtórne (metabolity kwasu arachidonowego, proteazy, wolne rodniki tlenowe, tlenek azotu) określane jako końcowe lub efektorowe, które są bezpośrednio odpowiedzialne za uszkodzenie funkcjonalne i strukturalne komórek [6,11,14].
Nadmiernie uwalniane mediatory wykazują właściwości wazoaktywne, przy czym mogą powodować miejscowy nieadekwatny skurcz lub rozkurcz łożyska naczyniowego, działają depresyjnie na mięsień sercowy, są supresorami układu odpornościowego, wywołują głębokie zaburzenia metabolizmu i prowadzą w efekcie do obumierania komórek, uszkodzenia narządów i MODS [6,11].
Ostatnie badania nad przebiegiem ogólnoustrojowej odpowiedzi zapalnej z określeniem roli mediatorów zapalenia w kształtowaniu obrazu klinicznego tego zespołu skłoniły Bone i wsp. [14] do uzupełnienia definicji przyjętych na konferencji uzgodnień ACCP/SCCM. Do dotychczasowej definicji zespołu SIRS proponuje się dodać kolejne dwa terminy: zespół kompensacyjnej reakcji przeciwzapalnej CARS ( compensatory anti-inflammatory syndrome) i mieszanej antagonistycznej reakcji MARS ( mixed antagonists response syndrome). Koncepcja ta oparta jest o wyniki badań nad aktywnością różnych grup cytokin zaangażowanych w reakcji zapalnej z uwzględnieniem ich przeciwstawnego pro- i antyzapalnego działania w zestawieniu z obserwowaną zmiennością objawów klinicznych [10,24].
Wprowadzenie do nazewnictwa terminu CARS zwróciło uwagę badaczy na wzajemne relacje między cytokinami pro- i przeciwzapalnymi jako wskaźnika dynamicznego stanu przywracania homeostazy ustrojowej. Przeciwzapalna aktywność w CARS wymierzona w hamowanie produkcji cytokin prozapalnych i odwracanie ich klinicznych skutków jest być może odpowiedzialna za fakt, że nie u wszystkich chorych obserwuje się przebieg kliniczny ciężkiego urazu lub zakażenia prowadzący do MODS. Może przyczyną narastania SIRS jest zaburzenie CARS? U niektórych chorych występuje przewaga reakcji przeciwzapalnej prowadząca w efekcie do hiporeaktywności, co zwiększa skłonność do ciężkich wtórnych powikłań infekcyjnych (ryc. 5).
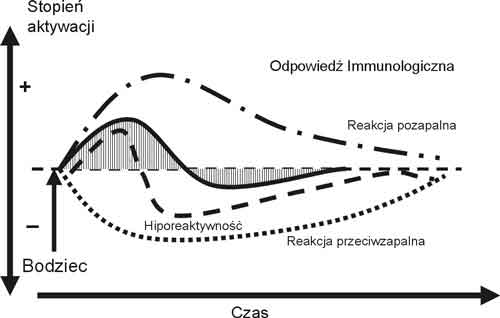
Ryc. 5. Schemat możliwych wariacji odpowiedzi zapalnej – pole zacienione przedstawia typowy przebieg aktywacji pro i przeciwzapalnej występujacy u większości chorych
Kompensacyjna reakcja przeciwzapalna może być szkodliwa i objawiać się klinicznie immunosupresją. Stan ten opisywany jako „ immunoparalysis ” lub „ window of immunodeficiency ” związany może być z zwiększoną podatnością chorych na zakażenia, co zostało pierwszy raz opisane w przebiegu leczenia ciężkich oparzeń. W tym okresie zwiększona jest liczba monocytów, ale o zmniejszonej ekspresji HLA-DR i HLA-DQ i zmniejszonej możliwości produkcji cytokin. Aktywność limfocytów T i B może być również hamowana przez endogenne glukokortykoidy i katecholaminy. Reakcja immunosupresji może być modulowana przez hamowanie przez IL-10 własnej produkcji, a także przez stymulację szpiku przez granulocyte-macrophage colony stumulating factor (GMCSF).
W najcięższym okresie SIRS związanym z niewydolnością wielonarządową MODS stan aktywności mediatorów zapalenia nazwać można immunologicznym dysonansem. Charakteryzuje się on nieprawidłowym i niezrównoważonym stanem immunologicznym z chaotyczną aktywacją całej sieci cytokin i nazwany został zespołem mieszanej antagonistycznej reakcji MARS. W obrazie klinicznym mogą dominować objawy ogólnoustrojowego zapalenia lub immunosupresji, a ryzyko zgonu w tym okresie jest wysokie i często związane ze stopniem aktywacji sieci cytokin [14,25,26,27].
Przedstawiona sekwencja przebiegu ogólnoustrojowej odpowiedzi zapalnej wyjaśnia udział w niej cytokin pro- i antyzapalnych i udokumentowana jest badaniami eksperymentalnymi, natomiast nie znajduje pełnego potwierdzenia w badaniach klinicznych. Produkcja sieci cytokin w przebiegu klinicznym posocznicy oraz po urazie jest bardzo zróżnicowana i zależna od wielu czynników obejmujących płeć, stan zdrowia poprzedzający incydent, utratę krwi, stan odżywienia a przede wszystkim rodzaj i siłę działania czynnika uszkadzającego. Ostatnie badania podkreślają ponadto genetycznie uwarunkowaną osobniczą zmienność w odpowiedzi immunologicznej na uraz i zakażenie. Zastanawiający jest fakt, że ogólnoustrojowe uwalnianie cytokin może wystąpić w wielu stanach chorobowych nie prowadząc wcale do niewydolności wielonarządowej [8,12,14,28,29,30].
Zastosowanie cytokin w diagnostyce SIRS
Trwają wciąż prace nad wyjaśnieniem złożonych procesów zachodzących w przebiegu SIRS i MODS. Prowadzone są liczne badania nad możliwościami wykorzystania wiedzy o roli cytokin w ogólnoustrojowej odpowiedzi zapalnej w diagnostyce ciężkich zaburzeń ogólnoustrojowych i w prognozowaniu wyników intensywnej terapii tych stanów. Poznanie roli cytokin w patomechanizmie ogólnoustrojowej reakcji zapalnej obudziło nadzieje na opracowanie skutecznej metody wczesnej diagnostyki tego stanu w oparciu o oznaczanie ich aktywności. Opracowano efektywne i dość dokładne metody oznaczania aktywności cytokin w krwi w oparciu o testy radioimmunologiczne (RIA) lub enzymatyczne (ELISA). Udokumentowano wzrost aktywności cytokin w stanie zapalnym. W oparciu o wartości stężeń endotoksyn (LPS) i wybranych cytokin zaproponowano nawet skalę prognostyczną w posocznicy (tab. II) [21].
Tab. II. Skala lipopolisacharydowo-cytokinowa ciężkości SIRS (LPS-Cytokine Score; wg 21)
| 0 | 2 | 4 |
| LPS (u ml-1) | < 0,2 | 0,2 - 2,0 | > 2,0 |
| TNF-alfa (pg ml-1) | < 40 | 40 - 80 | > 80 |
| Il-1 (pg ml-1) | < 40 | 40 - 200 | > 200 |
| IL-6 (pg ml-1) | < 250 | 250 - 500 | > 500 |
Kliniczne zastosowanie oznaczania stężeń cytokin w immunodiagnostyce SIRS ograniczone jest bardzo krótkim okresem półtrwania tych białek. Z tego powodu pierwotne mediatory reakcji zapalnej: TNF-alfa i interleukina-1, które inicjują tę odpowiedź i których wzrost aktywności potwierdzono w badaniach eksperymentalnych, nie znalazły szerokiego zastosowania w diagnostyce klinicznej. Większość badań klinicznych opiera diagnostykę o badanie aktywności interleukiny-6 jako wskaźnika stopnia aktywacji odpowiedzi zapalnej [31,32].
Badania ostatnich lat wykazały istotną rolę IL-6 w patogenezie różnych chorób o podłożu urazowym, infekcyjnym i zapalnym. Jej produkcja jest stymulowana przez IL-1 i TNF-alfa, a spadkowi aktywności tych cytokin towarzyszy spadek stężenia IL-6. Podanie IL-6 ochotnikom nie powoduje zaburzeń krążenia niezależnie od dawki [34,35].
Badania eksperymentalne wykazały, że IL-6 uwalniana jest gwałtownie ok. 60 minut po urazie a podwyższone jej stężenie stwierdza się w krwi obwodowej 12 – 48 godzin po urazie. Obecność interleukiny-6 w krwi obwodowej można wykazać u większości pacjentów z posocznicą a wartość jej stężenia może stanowić wskaźnik prognostyczny przebiegu choroby [13,14,24,34,35,43].
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Baue A.E., Durham R.: Systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndrome (MODS), multiple organ failure (MOF): are we winning the battle? Shock 1998, 10, 79-89.
2. Brun-Buisson C.: The epidemiology of systemic inflammatory response. Intensive Care Medicine 2000, 26, 864-874.
3. Glauser M.P.: Pathophysiologic basis of sepsis: considerations for future strategies of intervention. Critical Care Medicine 2000, 28, Suppl., S4-S8.
4. Salvo I., de Cian W., Musico M., Langer M., Piadena R., Wolfler A., Montant C., Magni E., the Sepsis Study Group: The Italian Sepsis study: Preliminary results on the incidence and evolution of SIRS, sepsis, severe sepsis and septic shock. Intensive Care Medicine 1995, 21, S244-S249.
5. Tran D.D., Groeneveld A.B., van der Meulen J., Nauta J.J., Strack van Schindel R.J., Thijs L.G.: Age, chronic disease, sepsis, organ system failure, and mortality in a medical intensive care unit. Critical Care Medicine 1990, 18, 474-479.
6. Schuster H.P.: Intensywna terapia w posocznicy, Sanmedica, Warszawa 1997.
7. Bartlett R.H.: Fizjologia stanów krytycznych, PZWL, Warszawa 1999.
8. Deutschman C.S.: Acute-phase response and SIRS/MODS: the good, the bad and the nebolous [review]. Critical Care Medicine 1998, 26, 1630-1631.
9. Reinhard K., Karzai W.: Indicators of the systemic response to severe infections. Acta Anaesthesiologica Scandinavica 1998, 4, Suppl, 133-135.
10. Robak T.: Biologia i farmakologia cytokin. Wydawnictwo Naukowe PZWN, Warszawa 1995.
11. Vincent J.L.: The immune response in critical illness: excessive, inadequate or dysregulated; in: The Immune Response in Critical Illness (Ed.: Marshall J.C., Cohen J.) Springer-Verlag, New York 1999.
12. Bion J.F.: Susceptibility to critical illness: reserve, response and therapy. Intensive Care Medicine 2000, 26, Suppl.1, S57-S63.
13. Dinarello C.C.A.: Proinflammatory and anti-inflammatory cytokines as mediators in the pathogenesis of septic shock. Chest 1997, 112, 321S-329S.
14. Bone R.C., Grodzin C.J., Balk R.A.: Sepsis: a new hypothesis for pathogenesis of the disease process. Chest 1997, 112, 235-243.
15. Calanda T., Heumann D.: Inhibitory cytokines; in: The Immune Response in Critical Illness (Ed.: Marshall J.C., Cohen J.) Springer-Verlag, New York 1999.
16. The ACCP/SCCM Consensus Conference Committee (1992) Definition for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992, 101, 1644-1655.
17. Haverman J.W., Kobold A.C., Tervaert J.W., van den Berg A.P., Tulleken J.E., Kallenberg C.G., The T.H.: The central role of monocytes in the pathogenesis of sepsis: consequences for immunomonitoring and treatment. Netherland Journal Medicine 1999, 55, 132-141.
18. Pinsky M.R.: Balancing the inflammatory response in sepsis; in: 1998 Yearbook of intensive care and emergency medicine (Ed.: Vincent J.L.) Springer-Verlag, New York 1998.
19. Talmor M., Hydo L.B.S., Barie P.S.: Relationship of systemic inflammatory response syndrome to organ dysfunction, length of stay and mortality in critical surgical illness: effect of intensive care unit resuscitaton. Archives of Surgery 1999, 134, 81-87.
20. Abraham E.: Why immunotherapies have not worked in sepsis. Intensive Care Medicine 1999, 25, 556-556.
21. Bone R.C.: Sepsis and its complications: the clinical problem. Critical Care Medicine 1994, 22, S8-S11.
22. Evans T.W., Smithies M.: ABC of intensive care: organ dysfunction. British Madical Journal 1999, 318, 1606-1609.
23. Faist E., Angele K., Zedler S.: Immunoregulation in shock, trauma and sepsis; in: The immune response in critical illness (Ed.: Marshall J.C., Cohen J.) 1999, Springer Verlag, New York, 312-334.
24. Muckart D.J., Bhagwanjee S.: American College of Chest Physicians/ Society of Critical Care Medicine Consensus Conference definitions of the systemic inflammatory response syndrome and allied disorders in relation to critically injured patients. Critical Care Medicine 1997, 25, 1789-1795.
25. Lyons A., Goebel A., Mannick J., Lederer J.: Protective effects of early interleukin-10 antagonism on injury-induced immune dysfunction. Archives of Surgery 1999, 134, 1317-1324.
26. Yang R.B., Mark M.R., Gray A., Huang A., Xie M.H., Zhang M., Goddard A., Wood W.I., Gurney A.L., Godowski P.J.: Toll-like receptor-2 mediates lipolysaccharide-induced cellular signaling. Nature 1998, 395, 931-937.
27. Ziegenfuss T., Wanner G.A., Grass C., Bauer I., Schuder G., Kleinshmidt S., Bauer M.: Mixed agonistic-antagonistic cytokine response in whole blood from patients undergoing abdominal aortic aneurysm repair. Intensive Care Medicine 1999, 25, 279-287.
28. Menges T., Boning O., Little Sg., Langefeld T., Welters I.: Genpolymorphisms of cytokines and serum proteases prognosis and outcome of trauma patients. Intensive Care Medicine 1999, 25, Suppl.1, S100 (259).
29. Eachempati S.R., Hydo L.R.N., Barie P.S.: Gender-based diferences in outcome in patients with sepsis. Archives of Surgery 1999, 134, 1342-1347.
30. Wichmann M.W., Inthorn D., Aandress H-J., Schildberg F.W.: Incidence and mortality of severe sepsis in surgical intensive care patients: the influence of patient gender on disease process and outcome. Intensive Care Medicine 2000, 26, 167-172.
31. Poeze M., Ramsay G.: Prognostic factors in pre-septic states; in: 1997 Yearbook of Intensive Care and Emergency Medicine (Ed.: Vincent J.L.), Springer Verlag, New York 1997.
32. Rhodes A., Newman P.J., Bennett E.D.: Prognostic markers in sepsis; in: 1998 Yearbook of Intensive Care and Emergency Medicine (Ed.: Vincent J.L.) Springer Verlag, New York 1998.
33. Kuhn P., Donato L., Coumaros G., Jernite M., Messer I.: Interleukin-6 (Il-6) and procalcitonin (PCT) as markers for the early diagnosis of neonatal bacterial infection. 15th Anual Meeting of ESPID 1997, abstract, 195.
34. Riad S., Bercker S., Ahlers O., Seiler S., Heidemann J., Falke K., Gerlach H.: Interleukin-6 and tumor necrosis factor in severely injured patients with multiple organ failure. Intensive Care Medicine 1999, 25, Suppl. 1, S100.
35. Van der Poll T., van Deventer S.J.H.: Interleukin-6 in bacterial infection and sepsis: innocent bystander or essential mediator? in: 1999 Yearbook of Intensive Care and Emergency Medicine (Ed.: Vincent JL) Springer Verlag, New York 1999.
36. Friedman G., Jankowski S., Marchant A., Goldman M.: Blood interleukin-10 levels paralell the severity of septic shock. Journal of Critical Care 1997, 12, 183-187.
37. Haupt W., Zirngibi H., Stehr A., Riese J., Holzheimer R.G., Hohenberger W.: Tumour necrosis factor-alpha and interleukin 10 production in septic patients and the regulatory effect of plasma. European Journal of Surgery 1999, 165, 95-100.
38. Tanigushi T., Koido Y., Aiboshi J., Yarnashita T., Suzaki S., Kurokawa A.: Change in the ratio of interlekin-6 to interleukin-10 predicts a poor outcome in patients with systemic inflammatory response syndrome. Critical Care Medicine 1999, 27, 1262-1264.
39. Kellum J.A., ALKarfy K.: Treating immunologic instability: a change in focus; in: 1999 Yearbook of Intensive Care and Emergency Medicine (Ed.: J.L. Vincent) Springer Verlag, New York 1999.
40. Kox W.J., Volk T., Kox S.N., Volk H-D.: Immunomodulatory therapies in sepsis. Intensive Care Medicine 2000, 26, Suppl.1, S124-S128.
41. Vincent J.L.: Search for effective immunomodulating strategies against sepsis. Lancet 1998, 351, 922-923.
42. Wheeler A.P., Bernard G.R.: Current concepts: treating patients with severe sepsis. New England Journal of Medicine 1999, 340, 207-214.
43. Barclay G.R.: Endotoxin-core antibodies: time for reappraisal? Intensive Care Medicine 1999, 25, 427-429.
44. Ahmed N., Marshall J.C.: Corticosteroid therapy in critical illness: a changing paradigm; in: 2000 Yearbook of Intensive Care and Emergency Medicine (Ed.: J.L. Vincent) Springer Verlag, New York 2000.
45. Lauterbach R., Zembala M.: Pentoxyphilline reduces plasma tumour necrosis factor-alpha concentration in premature infants with sepsis. Europhean Journal of Pediatry 1996, 155, 404-409.
46. Vary T.C.: Amrinone prevents the inhibition of muscle pyruvate dehydrogenase complex activity during sepsis. Shock 1996, 5, 229-232.
47. Bernard G.R., Wheeler A.P., Russel J.A.: The effects of ibuprofen on the physiology and survival of patients with sepsis. The ibuprofen in sepsis study group. New England Journal of Medicine 1997, 336, 952-953.
48. Panacek E., Marshall J., Fischkoff S.: Neutralization of TNF by monoclonal antibody improves survival and reduces organ dysfunction in human sepsis: results of the MONARC trial. Program and abstract of CHEST 2000: 66th Annual Scientific Assembly of the American College of Chest Physicians and Clinical World Congress on Diseases of the Chest; October 22-26, 2000; San Francisco. California.
49. Sprung C.L., Finch R.G., Thijs L.G., Glauser M.P.: International sepsis trial (INTERSEPT): role and impact of a clinical evaluation committee. Critical Care Medicine 1996, 24, 1441-1447.
50. De Vriese A.S., Vanholder R.C., Pascual M., Lameire N.H., Colardyn F.A.: Can inflammatory cytokines be removed efficiently by continous renal replacement therapies? Intensive Care Medicine 1999, 25, 903-910.