© Borgis - Postępy Nauk Medycznych 11/2012, s. 889-894
*Agnieszka Baranowska-Bik, Wojciech Zgliczyński
Zespół Cushinga a choroby układu sercowo-naczyniowego
Cushing’s syndrome and diseases of cardiovascular system
Klinika Endokrynologii Centrum Medycznego Kształcenia Podyplomowego, Warszawa
Kierownik Kliniki: prof. dr hab. med. Wojciech Zgliczyński
Streszczenie
Przewlekła hiperkortyzolemia związana jest z licznymi powikłaniami ze strony układu sercowo-naczyniowego. Badania populacyjne wskazują na zwiększoną śmiertelność w grupie pacjentów z zespołem Cushinga. Głównymi przyczynami śmiertelności są: choroba niedokrwienna serca, niewydolność serca, choroba zakrzepowo-zatorowa. W artykule przedstawiono patomechanizmy prowadzące do zaburzeń metabolicznych i schorzeń układu krążenia towarzyszących hiperkortyzolemii.
Summary
Chronic hipercortisolism is characterized by a series of serious complications involving cardiovascular system. Population-based studies indicate that Cushing’s syndrome is associated with increased mortality rate. Coronary artery disease, congestive heart failure and thromboembolic complications are among the main causes of death. In this review we present pathomechanisms of metabolic and cardiac complications in Cushing’s syndrome.

WSTĘP
Nadmierna produkcja kortyzolu występująca w zespole Cushinga prowadzi do szeregu zmian ogólnoustrojowych wywołujących w konsekwencji redystrybucję tkanki tłuszczowej, nadciśnienie tętnicze, zaburzenia gospodarki węglowodanowej, dyslipidemię oraz koagulopatie. Wszystkie wymienione patologie zwiększają ryzyko wystąpienia epizodów sercowo-naczyniowych nawet czterokrotnie. Badania populacyjne wskazują na zwiększoną śmiertelność w grupie pacjentów z zespołem Cushinga. Połowa nieleczonych umiera z powodu powikłań hiperkortyzolemii w ciągu 5 lat trwania choroby. Wśród głównych przyczyn śmiertelności dominują choroby układu sercowo--naczyniowego, w tym choroba niedokrwienna serca, niewydolność serca, choroba zakrzepowo-zatorowa (1, 2). Wpływ powikłań hiperkortyzolemii na rozwój chorób układu sercowo-naczyniowego przedstawiono na rycinie 1.
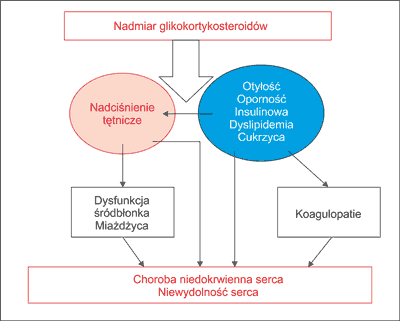
Ryc. 1. Wpływ powikłań hiperkortyzolemii na rozwój chorób układu sercowo-naczyniowego.
ZMIANY STRUKTURALNE I FUNKCJONALNE MIĘŚNIA SERCOWEGO W ZESPOLE CUSHINGA
U chorych z zespołem Cushinga obserwuje się zwiększenie masy lewej komory, często współistniejące z pogrubieniem przegrody międzykomorowej i koncentrycznym przerostem mięśnia serca oraz jego remodelingiem. Jednocześnie często dochodzi do upośledzenia funkcji lewej komory zarówno skurczowej, jak i rozkurczowej. Przerost mięśnia lewej komory wiąże się z niedokrwieniem miokardium oraz potencjalnie jest czynnikiem proarytmogennym, jak również sprzyjającym wystąpieniu niewydolności serca. Koncentryczny remodeling lewej komory zwiększa ryzyko wystąpienia zawału serca, niewydolności krążenia oraz udaru mózgu (2).
Wyniki badania prospektywnego prowadzonego przez Toja i wsp. na grupie pacjentów z zespołem Cushinga wskazują, że po uzyskaniu normalizacji poziomów kortyzolu w wyniku leczenia radykalnego, u większości chorych dochodzi do wycofania się zmian strukturalnych mięśnia serca; przede wszystkim przerostu i zwiększonej masy lewej komory, obserwowanych uprzednio w badaniach echokardiograficznych (2). Wyniki te są zgodne z pracą Pereiry i wsp., którzy również wykazali po skutecznym leczeniu hiperkortyzolemii regresję zmian strukturalnych lewej komory serca niezależnie od zmian ciśnienia tętniczego (3). W kilku badaniach obserwowano także pozytywne zmiany w badaniu echokardiograficznym, które występują w ciągu 4-18 miesięcy od uzyskania normokortyzolemii (3, 4, 5). Należy podkreślić, że zarówno poprawa struktury mięśnia serca, jak i czynności elektrycznej w wyniku ustąpienia hiperkortyzolemii prowadzi do zmniejszenia ryzyka wystąpienia epizodów sercowo-naczyniowych.
Nadciśnienie tętnicze jest jednym z głównych przyczyn przerostu lewej komory oraz jest udowodnionym czynnikiem pogarszającym wskaźniki sercowe u chorych z zespołem Cushinga, również u tych pacjentów, u których uzyskano wyleczenie z choroby podstawowej. Uważa się, że współistnienie hiperkortyzolemii i nadciśnienia tętniczego nasila ryzyko wzrostu masy mięśnia serca. Dowodem na to jest fakt, że chorzy wyleczeni z zespołu Cushinga, u których utrzymuje się nadciśnienie tętnicze mają większą częstość występowania nieprawidłowości w zakresie przebudowy mięśnia serca niż osoby z nadciśnieniem tętniczym bez hiperkortyzolemii w wywiadzie. Z drugiej strony, zmiany struktury lewej komory obserwowane są u osób z zespołem Cushinga bez nadciśnienia tętniczego, co może świadczyć o bezpośrednim wpływie kortyzolu na morfologię serca. Sugeruje się, że zmiany dotyczące lewej komory serca są częściowo związane z brakiem nocnego spadku ciśnienia tętniczego, zjawiskiem występującym u chorych z zespołem Cushinga (3).
Niekorzystne zmiany morfologiczne lewej komory serca mogą współistnieć z zaburzeniami funkcji skurczowej i rozkurczowej. W badaniach przeprowadzonych przez Toja i wsp. frakcja wyrzutowa nie różniła się znamiennie pomiędzy osobami z zespołem Cushinga a osobami zdrowymi. Jedynie u 20% badanych chorych występowały nieznaczne deficyty w zakresie funkcji skurczowej, ulegające remisji wraz z uzyskaniem prawidłowych stężeń kortyzolu. Autorzy wykazali prawidłową funkcję rozkurczową u 90% badanych (3). Jednakże w piśmiennictwie istnieją kontrowersje dotyczące zaburzeń funkcji rozkurczowej w przebiegu zespołu Cushinga wskazujące na upośledzenie rozkurczu u osób z niewydolnością serca (NYHA II i więcej) jak również w grupie pacjentów z wysokimi wartościami ciśnienia tętniczego lub źle kontrolowanym nadciśnieniem tętniczym (6-9).
NADCIŚNIENIE TĘTNICZE W ZESPOLE CUSHINGA
Szacuje się, że nadciśnienie tętnicze występuje u około 70-80% dorosłych pacjentów z zespołem Cushinga i u około 50% dzieci i młodzieży dotkniętych hiperkortyzolemią (1). Dodatkowo występują zaburzenia dobowego rytmu ciśnienia tętniczego z brakiem typowego spadku ciśnienia w godzinach nocnych (8). Jest to dodatkowy czynnik obciążający, zwiększający uszkodzenie narządów wewnętrznych i wpływający na śmiertelność (1).
Nadciśnienie tętnicze w przebiegu hiperkortyzolemii jest uwarunkowane wieloczynnikowo. Patomechanizm przedstawiono w tabeli 1.
Tabela 1. Patomechanizm nadciśnienia tętniczego w zespole Cushinga (10).
| Patomechanizm nadciśnienia tętniczego w zespole Cushinga |
Zahamowanie aktywności układu wazodylatacyjnego
Aktywność mineralokortykoidowa glikokortykoidów
Aktywacja układu renina-angiotensyna
Zwiększenie reaktywności układu sercowo-naczyniowego na działanie czynników naczynioskurczowych
Zwiększenie wrażliwości receptorów β-adrenergicznych na działanie katecholamin
Zahamowanie obwodowego rozkładu katecholamin gł. norepinefryny
↑ rzut serca, ↑ całkowity opór obwodowy, ↑ opór naczyń nerkowych |
Zahamowanie aktywności układu wazodylatacyjnego
Uznaje się, że glikokortykoidy hamują syntazę tlenku azotu prowadząc do zmniejszenia obwodowego rozkurczu naczyniowego. Dodatkowo wpływają również hamująco na produkcję prostacyklin oraz zmniejszają stężenia kalikreiny, kininy i prostaglandyny (PGE2) (10).
Aktywność mineralokortykoidowa glikokortykoidów
Glikokortykoidy mogą łączyć się zarówno z receptorem glikokortykoidowym, jak i mineralokortykoidowym (MR). Pomimo, że w warunkach fizjologicznych osoczowe stężenia kortyzolu są 100-1000 x wyższe niż stężenia aldosteronu to mechanizmy adaptacyjne z udziałem enzymu 11β dehydrogenazy hydroksysteroidowej typu 2 (11β-HSD 2) zapobiegają przyłączaniu kortyzolu do receptora mineralokortykoidowego. Enzym 11β-HSD 2, znajdujący się głównie w korze nerek, ale również w komórkach endotelium i mięśni gładkich naczyń, przekształca kortyzol w kortyzon, który nie łączy się z receptorem mineralokortykoidowym. W warunkach patologicznych nadmiaru kortyzolu dochodzi do przekroczenia zdolności wiążących enzymu 11β-HSD 2 i kortyzol wiążąc się z receptorem dla mineralokortykoidów naśladuje działanie aldosteronu wywołując zwiększoną reabsorcję sodu w kanalikach nerkowych i w konsekwencji wzrost objętości wewnątrznaczyniowej (10).
Aktywacja układu renina-angiotensyna
Glikokortykoidy wpływają na układ renina-angiotensyna przez zwiększanie aktywności angiotensynogenu. U chorych z zespołem Cushinga wykazano wzmożoną odpowiedź skurczową na angiotensynę II oraz spotęgowane centralne działanie protensyjne angiotensyny II (10).
Zwiększenie reaktywności układu sercowo-naczyniowego na działanie czynników naczynioskurczowych
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. De Leo M, Pivonello R, Auriemma RS et al.: Cardiovascular disease in Cushing’s syndrome: heart versus vasculature. Neuroendocrinology 2010; 92 (Suppl. 1): 50-54.
2. Toja PM, Branzi G, Ciambellotti F et al.: Clinical relevance of cardiac structure and function abnormalities in patients with Cushing’s syndrome before and after cure. Clin Endocrinol (Oxf) 2012; 76(3): 332-338.
3. Pereira AM, Delgado V, Romijn JA et al.: Cardiac dysfunction is reversed upon successful treatment of Cushing’s syndrome. Eur J Endocrinol 2010; 162(2): 331-340.
4. Sugihara N, Shimizu M, Kita Y et al.: Cardiac characteristics and postoperative courses in Cushing’s syndrome. Am J Cardiol 1992; 69(17): 1475-1480.
5. Fallo F, Budano S, Sonino N, Muiesan et al.: Left ventricular structural characteristics in Cushing’s syndrome. J Hum Hypertens 1994; 8(7): 509-513.
6. Takagi S, Tanabe A, Tsuiki M et al.: Hypokalemia, diabetes mellitus, and hypercortisolemia are the major contributing factors to cardiac dysfunction in adrenal Cushing’s syndrome. Endocr J 2009; 56(8): 1009-1018.
7. Baykan M, Erem C, Gedikli O et al.: Assessment of left ventricular diastolic function and Tei index by tissue Doppler imaging in patients with Cushing’s Syndrome. Echocardiography 2008; 25(2): 182-190.
8. Muiesan ML, Lupia M, Salvetti M et al.: Left ventricular structural and functional characteristics in Cushing’s syndrome. J Am Coll Cardiol 2003; 41(12): 2275-9.
9. Fallo F, Maffei P, Dalla Pozza A, Carli M et al.: Cardiovascular autonomic function in Cushing’s syndrome. J Endocrinol Invest 2009; 32(1): 41-45.
10. Cicala MV, Mantero F: Hypertension in Cushing’s syndrome: from pathogenesis to treatment. Neuroendocrinology 2010; 92 (Suppl. 1): 44-49.
11. Faggiano A, Pivonello R, Spiezia S et al.: Cardiovascular risk factors and common carotid artery caliber and stiffness in patients with Cushing’s disease during active disease and 1 year after disease remission. J Clin Endocrinol Metab 2003; 88(6): 2527-2533.
12. Albiger N, Testa RM, Almoto B et al.: Patients with Cushing’s syndrome have increased intimal media thickness at different vascular levels: comparison with a population matched for similar cardiovascular risk factors. Horm Metab Res 2006; 38(6): 405-410.
13. Fallo F, Famoso G, Capizzi D et al.: Coronary microvascular function in patients with Cushing’s syndrome. Endocrine 2012; DOI: 10.1007/s12020-012-9764-9762.
14. Trementino L, Arnaldi G, Appolloni G et al.: Coagulopathy in Cushing’s syndrome. Neuroendocrinology 2010; 92 (Suppl. 1): 55-59.
15. Casonato A, Pontara E, Boscaro M et al.: Abnormalities of von Willebrand factor are also part of the prothrombotic state of Cushing’s syndrome. Blood Coagul Fibrinolysis 1999; 10(3): 145-151.
16. Erem C, Nuhoglu I, Yilmaz M et al.: Blood coagulation and fibrinolysis in patients with Cushing’s syndrome: increased plasminogen activator inhibitor-1, decreased tissue factor pathway inhibitor, and unchanged thrombin-activatable fibrinolysis inhibitor levels. J Endocrinol Invest 2009; 32(2): 169-174.
17. Faggiano A, Melis D, Alfieri R et al.: Sulfur amino acids in Cushing’s disease: insight in homocysteine and taurine levels in patients with active and cured disease. J Clin Endocrinol Metab 2005; 90(12): 6616-6622.
18. Tsuiki M, Tanabe A, Takagi S et al.: Cardiovascular risks and their long-term clinical outcome in patients with subclinical Cushing’s syndrome. Endocr J 2008; 55(4): 737-745.
19. Fallo F, Sonino N: Should we evaluate for cardiovascular disease in patients with Cushing’s syndrome? Clin Endocrinol (Oxf) 2009; 71(6): 768-71.
20. Chanson P, Salenave S: Metabolic syndrome in Cushing’s syndrome. Neuroendocrinology 2010; 92 (Suppl. 1): 96-101.
21. Arnaldi G, Scandali VM, Trementino L et al.: Pathophysiology of dyslipidemia in Cushing’s syndrome. Neuroendocrinology 2010; 92 (Suppl. 1): 86-90.
22. Valassi E, Biller BM, Klibanski A, Misra M: Adipokines and cardiovascular risk in Cushing’s syndrome. Neuroendocrinology 2012; 95(3): 187-206.
