© Borgis - Nowa Stomatologia 1-2/2009, s. 40-44
Monika Kozarzewska1, Anna Matosek1, *Dorota Olczak-Kowalczyk1, 2, Anna Marczak-Hałupka3, Barbara Adamowicz-Klepalska4, Krystyna H. Chrzanowska5
Objawy kliniczne w jamie ustnej w zespole Silvera i Russella. Opis przypadku
Clinical manifestation in oral cavity in Silver-Russell syndrome. Case report
1Zakład Patologii Jamy Ustnej Instytutu „Pomnik-Centrum Zdrowia Dziecka”, Warszawa
Kierownik Zakładu: dr hab. n. med. Dorota Olczak-Kowalczyk
2Zakład Stomatologii Dziecięcej Instytutu Stomatologii Warszawskiego Uniwersytetu Medycznego
Kierownik Zakładu: dr n. med. Aleksander Remiszewski
3Poradnia Genetyczna Wojewódzkiego Zespołu Ochrony Zdrowia Matki, Dziecka i Młodzieży, Katowice
Kierownik Poradni: lek. med. Marek Labus
4Katedra i Zakład Stomatologii Wieku Rozwojowego Akademii Medycznej w Gdańsku
Kierownik Katedry i Zakładu: prof. dr hab. n. med. Barbara Adamowicz-Klepalska
5Zakład Genetyki Medycznej Instytutu „Pomnik-Centrum Zdrowia Dziecka”, Warszawa
Kierownik Zakładu: prof. dr hab. n. med. Małgorzata Krajewska-Walasek

WSTĘP
Zespół Silvera i Russella (Silver-Russell syndrome; SRS; OMIM 180860) jest rzadkim, klinicznie i genetycznie heterogennym zespołem wad rozwojowych (1, 2). Po raz pierwszy został opisany niezależnie przez Silvera i wsp. w 1953 oraz Russella w 1954 roku. Nazwa zespołu jest eponimem utworzonym od nazwisk autorów pierwszych opisów.
Częstość występowania zespołu określa się na 1:30 000 do 1:100 000, jednakowo często rozpoznawany jest u obu płci (2). Dla SRS charakterystyczna jest ciężka dystrofia wewnątrzmaciczna (ang. intrauterine growth retardation; IUGR), słaby przyrost wzrostu i masy ciała po urodzeniu, względna makrocefalia (pseudowodogłowie), charakterystyczna trójkątna twarz (duże czoło, niedorozwój części środkowej i mała żuchwa) oraz asymetria, która może dotyczyć twarzy, kończyn i/lub tułowia. W okresie wczesnodziecięcym występują niedobory tkanki podskórnej, tłuszczowej, zmniejszenie napięcia mięśniowego (1, 2, 3, 4, 5, 6, 7, 8). Występowaniu zmian morfologicznych towarzyszą zaburzenia ogólnoustrojowe. U pacjentów z zespołem Silvera i Russella często obserwowano wielokrotne infekcje ucha środkowego prowadzące do pogorszenia, a nawet utraty słuchu.
Wśród innych objawów, występujących z różną częstością, wymienia się m.in.: problemy ze ssaniem i/lub połykaniem, brak uczucia głodu utrzymujący się do około 3. roku życia, opóźniony rozwój psychoruchowy, a także występowanie różnych anomalii rozwojowych, dotyczących m.in. uzębienia. W jamie ustnej obserwowano zwężenie łuków zębowych, prowadzące do zaburzeń zgryzowo-zwarciowych (1, 2, 3, 9). Mikrognację opisywano u około 50% pacjentów (10, 11). Najczęściej występującymi nieprawidłowościami zgryzowymi były stłoczenia zębów oraz wady z grupy tyłozgryzów. Ponadto u pacjentów z zespołem Silvera-Russella opisywano zaburzenia kształtu i wielkości zębów, podniebienie gotyckie oraz hipodoncję dotyczącą zębów przedtrzonowych (1, 2, 9, 12).
W piśmiennictwie dostępne są także informacje na temat występowania u pacjentów z SRS nowotworów wywodzących się z komórek wątroby, guzów jąder oraz craniopharyngoma (2). Nie stwierdzono jednak żadnych zależności pomiędzy tym zespołem a rozwojem procesów nowotworowych.
Zespół Silvera i Russella z reguły występuje sporadycznie, natomiast od początku podejrzewano udział czynników genetycznych w patogenezie choroby i sugerowano, że heterogenność kliniczna może być odzwierciedleniem heterogenności genetycznej. Zasadniczą rolę w patogenezie choroby odgrywają mechanizmy epigenetyczne (dziedziczenie epigenetyczne). Dotąd udaje się wyjaśnić podłoże około 45-70% przypadków, w których podejrzewany jest SRS (13). Disomia matczynej kopii chromosomu 7 odpowiada za cechy charakterystyczne dla SRS u 5-10% pacjentów (14), natomiast hipometylacja w regionie p15 na matczynej kopii chromosomu 11, w którym znajduje się piętnowany zespół genów IGF2-H19mających kluczowe znaczenie dla kontroli wzrostu, rozpoznawana jest u około 35-65% dzieci z fenotypem SRS (15). Ponadto u około 1% dzieci stwierdza się małe aberracje strukturalne różnych chromosomów (m.in. 1, 7, 11, 15 i 17 pary) (1). W pozostałych przypadkach patogeneza zaburzeń pozostaje nadal niejasna.
Celem pracy jest prezentacja cech fenotypowych zespołu Silvera i Russella u 10-letniego chłopca.
OPIS PRZYPADKU
10-letni chłopiec został skierowany do Zakładu Patologii Jamy Ustnej z Poradni Genetycznej „Instytutu Pomnik-Centrum Zdrowia Dziecka” w trakcie badań diagnostycznych w kierunku Zespołu Silvera-Russella, w celu oceny zaburzeń w układzie stomatognatycznym.
Na podstawie wywiadu ustalono, że dziecko pochodzi z pierwszej ciąży o prawidłowym przebiegu, rozwiązanej w 38 tygodniu cięciem cesarskim z powodu poprzecznego ułożenia płodu. Stan noworodka z masą urodzeniową 1350 g określono w pierwszej minucie na 7 punktów, w trzeciej minucie na 8 punktów wg skali Apgar. Długość ciała dziecka wynosiła 37 cm, obwód głowy 31 cm, obwód klatki piersiowej 32 cm. W badaniu klinicznym po urodzeniu stwierdzono obniżenie napięcia mięśniowego noworodka, znaczne upośledzenie wzrostu oraz niedobór masy ciała. Ponadto zauważono cechy dysmorfii, dużą mózgoczaszkę, nieproporcjonalnie długą i szeroką przy prawidłowym wymiarze, głęboko osadzone gałki oczne, płaskie nisko osadzone małżowiny uszne, trójkątną twarzoczaszkę, mikrostomię. Stwierdzono asymetrię kończyn (tab. 1). Obserwowano także opóźniony rozwój psychoruchowy oraz opóźniony rozwój mowy (wymaga pomocy logopedy).
Tabela 1. Cechy stwierdzone u chłopca z zespołem Silvera i Russella bezpośrednio po urodzeniu.
| Płeć | M |
| Niska masa urodzeniowa (Ł3 centyla) | 1350 g
(38 hbd) |
| Obwód głowy przy urodzeniu | 31 cm |
| Zahamowanie wzrastania po urodzeniu (wzrost Ł3 centyla) | + |
| Względna makrocefalia (obwód głowy odp. do wieku) | + |
| Charakterystyczna twarz* | + |
| Asymetria twarzy/ciała i/lub kończyn | + |
| Klinodaktylia V palców | + |
| Spodziectwo | + |
*Dysproporcja twarzowo-czaszkowa (mała twarzoczaszka), twarz mała, trójkątna, wysokie czoło, wydatne guzy czołowe, kąciki ust skierowane do dołu, mała bródka.
W badaniu klinicznym w wieku 10 lat zwrócił uwagę niedobór wzrostu i masy ciała (pacjent został zakwalifikowany do terapii hormonem wzrostu) oraz pogłębiająca się asymetria kończyn, uboga tkanka podskórna.
Zespół Silvera i Russella rozpoznano na podstawie obrazu klinicznego. Badania genetyczne wykazały prawidłowy kariotyp 46XY oraz prawidłowe oburodzicielskie dziedziczenie chromosomów 7 i 14 pary (14).
W badaniu stomatologicznym zewnątrzustnym stwierdzono małą, asymetryczną twarz z wysokim czołem, wydatnymi guzami czołowymi, trójkątny kształt dolnego piętra twarzy, kąty ust skierowane do dołu oraz małą bródkę (ryc. 1). Nie wykazano nieprawidłowości czynnościowych stawów skroniowo-żuchwowych.
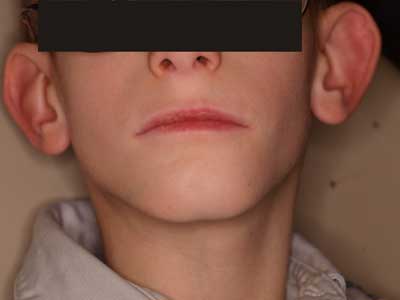
Ryc. 1. Asymetria i trójkątny kształt dolnego piętra twarzy, kąty ust skierowane do dołu, mała bródka oraz odstające i nisko osadzone małżowiny uszne u 10-letniego pacjenta z zespołem Silvera i Russella.
W badaniu wewnątrzustnym zaobserwowano zwężenie górnego i dolnego łuku zębowego, podniebienie wąskie i wysoko wysklepione oraz uzębienie mieszane (ryc. 2a). Analiza kliniczna warunków zgryzowo-zwarciowych wykazała cechy wady dotylnej – tyłozgryzu.
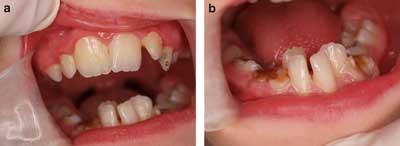
Ryc. 2. Zwężenie obu łuków zębowych, nieprawidłowe ustawienie zębów i choroba próchnicowa u 10-letniego chłopca z zespołem Silvera i Russella.
a) Przechylenie lewego zęba siecznego szczęki w kierunku przedsionka ( protrusio).
b) Nieprawidłowe ustawienie zębów siecznych bocznych żuchwy ( infrapositio).
W obrębie uzębienia stwierdzono zęby o prawidłowym kształcie bez zaburzeń rozwojowych szkliwa, przechylenie lewego zęba siecznego szczęki w kierunku przedsionka ( protrusio), nieprawidłowe ustawienie zębów siecznych bocznych żuchwy ( infrapositio) oraz obecność licznych ubytków próchnicowych o różnym stopniu zaawansowania w obrębie 4 zębów stałych (16, 26, 36, 65) oraz zębów mlecznych (55, 63, 64, 75, 74, 73, 83, 84, 85) (ryc. 2a i b).
Stan higieny jamy ustnej oceniono jako dostateczny (OHI=1,5). Stwierdzono także objawy umiarkowanego zapalenia dziąseł dotyczące dziąsła brzeżnego i brodawek dziąsłowych (GI=1,46). Błona śluzowa jamy ustnej była prawidłowo zabarwiona, wilgotna i napięta, bez zmian patologicznych.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Midro AT, Zajączek S: Zespół Silvera i Russella. Przyczyny powstawania, diagnoza i elementy poradnictwa genetycznego. Przegl Pediat 2007; 37(4): 393-400. 2. Zajączek S: Zespół Silvera-Russella: wyzwanie dla genetyki i pediatrii. Klin Pediatr 2002; 10: 431-5. 3. Hubert E, Midro AT: Syndromic retro and/or micrognathism of mandible in different genetic entities. J Appl Genet 1998; 39A: 68. 4. Donnai Det al.: Severe Silver-Russell syndrome. J Med Genet 1989; 26: 447-51. 5. Escobar V, Gleiser S, Weaver DD: Phenotypic and genetic analysis of the Silver-Russell syndrome. Clin Genet 1978; 13: 278-88. 6. Midro AT, Rogowska M: Interdyscyplinarne postępowanie diagnostyczne w przypadku wykrycia aberracji chromosomowych u pacjentów z upośledzeniem umysłowym. Biuletyn STG 1995; 14: 26-33. 7. Rybakowa M, Sokołowski J: Wczesny i późny obraz kliniczny karłowatości typu Silver-Russell. Pediatr Pol 1974; 47: 207-11. 8. Sachnowska K i wsp.: Zespół Silvera-Russella, jedna z postaci wewnątrzmacicznego zahamowania rozwoju na podstawie własnych przypadków i przeglądu literatury. Endokrynol Pol 1981; 32: 137-52. 9. Wollman HA et al.: Growth and symptoms in Silver-Russell syndrome: rewiev on the basis of 386 patients. Eur J Pediatr 1995; 154: 958-68. 10. Lewandowska K, Rymkiewicz-Kluczyńska B: Obraz kliniczny i efekty leczenia hormonem wzrostu dzieci z zespołem Silvera-Russella. Pediatr Pol 2002; LXXVII (7): 599-606. 11. Price SM et al.: The spectrum of Silver-Russell syndrome: a clinincal and molecular genetic study and new diagnostic criteria. J Med Genet 1999; 36(11): 837-42. 12. Abraham E, Altiok H, Lubicky JP: Musculoskeletal manifestations of the Silver-Russell syndrome. J Pediat Orthop 2004; 24: 552-64. 13. Abu-Amero S et al.: The genetic etiology of Silver-Russell syndrome. J Med Genet 2008; 45: 193-9. 14. Kotzot D et al.: Maternal uniparental disomy - review and further delineationof the phenotype. Eur J Pediatr 2000, 159: 247-56. 15. Gicquel C, Rossignol S, Cabrol S: Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet 2005; 37: 1003-7. 16. Szpringer-Nodzak M: Ząbkowanie. [W:] Springer-Nodzak M, Wochna-Sobańska M: Stomatologia Wieku Rozwojowego, Wydawnictwo Lekarskie PZWL, Warszawa; 1999: 40-54. 17. Bartholdi D et al.: Epigenetic mutations of the imprinted IGF2-H19 domain in Silver-Russell syndrome (SRS): results from a large cohort of patients with STS and SRS-like phenotypes. J Med Genet 2009; 46: 192-7. 18. Midro AT i wsp.: Zespół Silvera i Russella u chłopca z obniżoną sprawnością umysłową. Pediatr Pol 1993; 68: 85-9. 19. Maszkiewicz W i wsp. Zespół Silvera-Russella. Post Neonatol 2000; 1: 360. 20. Sobkowiak E i wsp. Somatic development of children who recovered from secondary malabsorption syndrome. Pol Merk Lek 2000; 8: 409-10. 21. Dinner M et al.: Anestetic implications. Anesth Analog 1994; 78: 1197-9. 22. Lai KY et al.: Cognitive abilitiesassociated with the Silver-Russel syndrome. Arch Is Child 1994; 71: 490-6.
