© Borgis - Nowa Pediatria 3/2005, s. 111-114
Mieczysław Chmielik, Anna Gabryszewska, Eliza Brożek
Ucisk oczodołu u 6-letniego chłopca z mukowiscydozą
Compression of the orbit in a 6-years old boy with cystic fibrosis
z Kliniki Otolaryngologii Dziecięcej Akademii Medycznej w Warszawie
Kierownik Kliniki: prof. dr hab. med. Mieczysław Chmielik
Streszczenie
Cystic fibrosis is one of most common genetically inherited, metabolic diseases connected with mutation of the gene coding a CFTR (cystic fibrosis transmembrane regulator) protein. In the ENT specialist practice it may appear in form of chronic sinusitis, mucocoele, mucopyocoele or polyps. While chronic sinusitis may occur at every age, mucocoele usually appear before the 5th year of age and polyps after the 4th year. The treatment is usually multifocal. A case of a boy with cystic fibrosis and mucopyocoele has been described in this paper.

WSTĘP
Mukowiscydoza jest najczęstszą chorobą metaboliczną uwarunkowaną genetycznie. Częstość występowania w Polsce wynosi 1:2300 żywo urodzonych niemowląt, a np. u Azjatów 1:90 000 żywych urodzeń. Jest to choroba dziedziczona autosomalnie recesywnie z różną penetracją genu. Gen zlokalizowany jest na długim ramieniu chromosomu 7 (3) i koduje białko pełniące rolę kanału chlorkowego zależnego od cAMP (CFTR – cystic fibrosis transmembrane regulator) (2, 8). Występuje ono w komórkach nabłonka dróg oddechowych, dróg żółciowych, gruczołów potowych, przewodów trzustkowych i w nasieniowodach. Zaburzenia funkcji wydzielania jonów chlorkowych prowadzą do zwiększania lepkości wydzieliny, a w konsekwencji do blokowania przewodów wyprowadzających. CFTR bierze udział w transporcie jonów chlorkowych poprzez inne niezależne od cAMP kanały jonowe. Ponadto w mukowiscydozie dochodzi do różnych zaburzeń komórkowych. Nieprawidłowa aktywność CFTR prowadzi do zmniejszenia ilości syntetazy dwutlenku azotu w komórkach nabłonka, co w konsekwencji powoduje spadek NO w tkankach i nasilenie odpowiedzi zapalnej i upośledzenie niszczenia bakterii; występują zaburzenia produkcji gangliozydów odpowiedzialnych m.in. za właściwości powierzchniowe komórek (np. zwiększa się ilość miejsc wiązania dla Pseudomonas aeruginosa); stwierdzono niedobór interleukiny – 10 w komórkach nabłonka chorych na mukowiscydozę (2).
Obecnie poznanych jest ponad 800 mutacji genu CFTR (2). Można je podzielić na silne (zagrażające życiu) i łagodne (powodujące lżejszy przebieg choroby). Najczęstszą mutacją występującą w 70% (w Polsce w 56%) przypadkach jest silna mutacja delta F508. Objawy w mukowiscydozie dotyczą zmian w obrębie:
– trzustki: zwyrodnienie torbielowate, zapalenie trzustki, zaburzenia wchłaniania, cukrzyca,
– wątroby: zastój żółci, marskość,
– jelit: niedrożność smółkowa, podniedrożności przewodu pokarmowego,
– nasieniowodów,
– płuc, dolnych dróg oddechowych: zapalenia przewlekłe, rozstrzenie, zaburzenia krążenia płucnego, serce płucne,
– górnych dróg oddechowych: przewlekłe zapalenie zatok (mucopyosinusitis), polipy nosa,
Rozpoznanie jest oparte na podstawie charakterystycznych zmian płucnych, zaburzeń trawienia oraz dodatniego testu potowego (norma zawartości jonów sodu w pocie wynosi Na+<60 mmol/l a jonów chlorokowych Cl–<50 mmol/l).
Możemy wyróżnić trzy główne postacie mukowiscydozy:
1. Postać płucną – dominują objawy ze strony dróg oddechowych.
2. Postać brzuszną – z objawami głównie ze strony przewodu pokarmowego.
3. Postać mieszaną – łączy objawy obydwu powyższych.
Leczenie mukowiscydozy jest wielokierunkowe i obejmuje antybiotykoterapię, fizjoterapię, suplementację enzymów trawiennych, stosowanie odpowiedniej diety, farmakoterapię, leczenie chirurgiczne. Prowadzone są badania nad terapią genową.
OPIS PRZYPADKU
Chłopiec urodzony z ciąży II, porodu I, na 10 punktów w skali Apgar, z masą urodzeniową 3500 g został przyjęty do Oddziału Otolaryngologii Dziecięcej w wieku 6 lat z powodu narastającej deformacji w obrębie oczodołu prawego, okresowo występujących bólów głowy, ropnych wycieków z jam nosa i miernie upośledzonej drożności nosa. W wieku 3 miesięcy u chłopca rozpoznano mukowiscydozę (postać mieszaną). Pozostawał pod stałą opieką pulmonologiczną i gastrologiczną. Wielokrotnie przebył zapalenie płuc i zapalenia oskrzeli, z powodu których wymagał hos-pitalizacji. Leczenie przy przyjęciu obejmowało: inhalacje z 0,9% NaCl z gentamycyną i pulmozyną, Biliepar, Kreon, Mucosolvan. Rok przed przyjęciem obserwowano zwężenie prawej szpary powiekowej, opadanie powieki górnej. W wykonanym wówczas badaniu NMR głowy i oczodołów stwierdzono: przerost błony śluzowej wszystkich zatok oraz w obrębie prawego oczodołu wpuklanie się rozdętej komórki sitowia modelującej i przemieszczającej mięsień okoruchowy przyśrodkowy. Nie stwierdzono zmian w obrębie mózgowia i skrzyżowania nerwów wzrokowych. Nie podjęto wówczas leczenia chirurgicznego.
Przy przyjęciu chłopiec był w stanie ogólnym dobrym, bez gorączki, bez cech zaostrzenia infekcji, z niedoborem masy poniżej 3 centyla.
W badaniu stwierdzono asymetrię szpar powiekowych (prawa węższa) w przyśrodkowo-górnym kwadrancie oczodołu prawego, dość twardy, niebolesny guzek o średnicy ok. 15 mm, poszerzenie nasady nosa. Ruchomość gałek ocznych była zachowana.
W rynoskopii przedniej uwidoczniono przerośniętą małżowinę nosową środkową, śluzówki różowe, nieznaczną ilość wydzieliny śluzowej w obu jamach nosa.
Badania laboratoryjne wykazały: podwyższone wartości OB (28 mm/h) i leukocytozę (14 000/mm3). Chłopca konsultowano okulistycznie. Pomimo przemieszczenia gałki ocznej prawej w stronę prawą i do dołu ruchy gałką oczną były zachowane, prawidłowe. W ocenie dna oczu stwierdzono zatarte granice tarcz od góry i dołu, obraz dna oczu mieścił się w granicach normy dla wieku. U pacjenta rozpoznawano wcześniej krótkowzroczność. Wada wzroku była skorygowana szkłami korekcyjnymi. Nie stwierdzono pogorszenia ostrości wzroku w badaniu okulistycznym.
W wykonanym badaniu tomograficznym zatok przynosowych i oczodołów wykonanym techniką spiralną w rekonstrukcjach co 3 mm stwierdzono masywne zmiany w obrębie wszystkich zatok, zmiany zanikowe kości ścian oczodołu prawego, przemieszczenie mięśnia przyśrodkowego i nerwu wzrokowego bocznie przez rozdęte komórki sitowia. Po stronie lewej rozdęta komórka sitowia uwypukliła boczną ścianę nosa, która dochodząc do przegrody nosa, przemieściła ją. Po stronie prawej rozdęta zatoka czołowa wpukla się w część przednią oczodołu. Nie stwierdzono w badaniu radiologicznym destrukcji kości ograniczających przedni dół czaszki (ryc. 1, 2, 3).
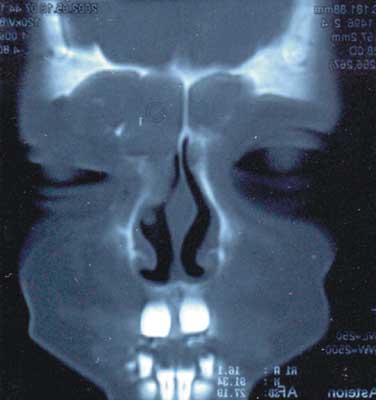
Ryc. 1. Tomografia komputerowa zatok przynosowych w płaszczyźnie wieńcowej – widoczna całkowita bezpowietrzność wszystkich zatok.
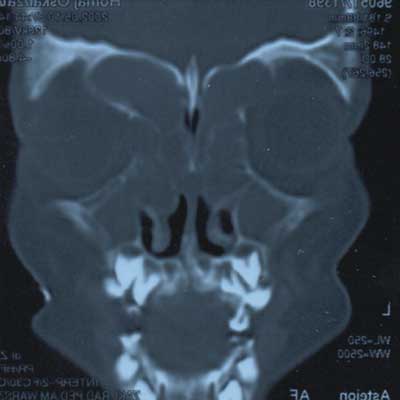
Ryc. 2. Obraz tomografii komputerowej przedstawiający rozdętą zatokę czołową, wpuklającą się w obręb oczodołu prawego.
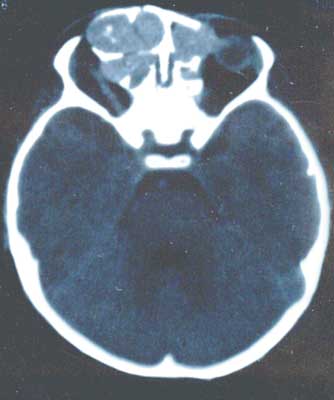
Ryc. 3. Tomografia komputerowa oczodołów z uwidocznieniem nerwu wzrokowego.
Chłopiec został zakwalifikowany do leczenia operacyjnego zatok przynosowych. Zaplanowano zabieg wykorzystując zarówno metody endoskopowe jak i otwarcie zatoki czołowej i sitowia z dojścia zewnętrznego. Osłonowo w okresie okołoperacyjnym włączono do leczenia Fortum.
W I etapie wykonano operację po stronie prawej. Podczas operacji stwierdzono ścieńczenie przedniej ściany zatoki czołowej, oraz blaszek w obrębie błędnika sitowego. Zatoki zawierały gęstą, maziową, zielonkawą treść ropną. Usunięto wyrostek haczykowaty i przerośniętą polipowato błonę śluzową z okolic ujścia zatok. Podobną treść odessano i wypłukano z zatoki szczękowej. W zatokach pozostawiono dreny (w zatoce czołowej dren zewnętrzny). Podobne postępowanie zabiegowe przeprowadzono po stronie lewej po upływie miesiąca.
W okresie pooperacyjnym przez 9 dni w I etapie i 7 dni w II etapie wykonywano codziennie płukanie zatok przynosowych solą fizjologiczną z antybiotykiem i mesną. Nie obserwowano powikłań po zabiegu, gojenie przebiegało prawidłowo. Po usunięciu drenów zalecono codzienne inhalacje z soli fizjologicznej z dodatkiem mukolityku i antybiotyku.
W posiewie wyhodowano zlewny wzrost Pseudomonas aeruginosa wrażliwego na zastosowany ceftazidym – kontynuowano antybiotykoterapię.
W badaniu histopatologicznym stwierdzono w obrębie błony śluzowej zatok nacieki zapalne głównie z komórek plazmatycznych i limfocytów.
Od czasu operacji do badania kontrolnego po 6 miesiącach, chłopiec był raz hospitalizowany z powodu zaostrzenia objawów ze strony dolnych dróg oddechowych. W dniu badania chłopiec w stanie ogólnym dobrym, bez dolegliwości, bez cech zaostrzenia infekcji. W badaniu kontrolnym w rynoskopii stwierdzono: zachowaną drożność jam nosa, przerośniętą małżowinę nosową środkową prawą, drożne ujścia zatok szczękowych, niewielką ilość wydzieliny powyżej małżowiny środkowej.
DYSKUSJA
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Brihaye P. et al.: Chronic rhinosinusitis in cystic fibrosis(mucoviscidosis). Acta oto-rhino-laryngologica belg.,1997, 51,323-337. 2. Pamela B. Davis, M.D.: Mukowiscydoza. Pediatria po Dyplomie, Luty 2002, Vol. 6, Nr 1,33-41. 3. Thome D.C. et al.: Bilateral ethmoidal mucocele in cystic fibrosis:report of a case. Inter J Ped Otorhinolaryngology. 2000, 55:143-148. 4. Brihaye P. et al.: Pathological changes of the lateral nasal wall in patients with cystic fibrosis (mucoviscidosis). Inter J Ped Otorhinolaryngology, 1994, 28:141-147. 5. Wiatrak B.J. M.D.: Cystic Fibrosis Presenting With Sinus Disease in Children. The pediatric Forum Mar 1993, Vol.147: 258-260. 6. Guttenplan M.D., Wetmore R.F.: Paranasal Sinus Mucocele in Cystic Fobrosis. Clinical Pediatrics, Sep 1989, Vol. 28 No. 9:429-430. 7. Lund V.J., Rolfe M.E.: Ophtalmic considerations in fronto-ethmoidal mucocoeles. The Journal of Laryngology and Otology. Jul 1989, Vol. 103:667-669. 8. Rowe-Jones J.M., Mackay I.S.: Endoscopic Sinus Surgery in the Treatment of Cystic Fibrosis With nasal Poliposis. Laryngoscope Dec 1996, 106:1540-43. 9. Coste A. et al.: Endoscopic and CT-scan evaluation of rhinosinusitis in cystic fibrosis. Rhinology 1995, 33:152-156. 10. Tunkel D.E. et al.: Bilateral maxillary sinus mucoceles in an infant with cystic fibrosis. Otolaryngology-Head and neck Surgery, Jul 1994; 111:116.
