© Borgis - Postępy Nauk Medycznych 4/2000, s. 60-63
Witold Kucharski, Andrzej Misiak
Ostre porfirie wątrobowe: rozpoznanie, leczenie, czynniki wywołujące atak choroby
Acute hepatic porphyrias: diagnosis, treatment, precipitating factors
Klinika Chirurgiczna, Poradnia i Pracownia dla Chorych na Porfirię, Instytut Hematologii i Transfuzjologii w Warszawie
Kierownik Kliniki: prof. dr hab. med. Jan Marek Ziemski
Kierownik Poradni: dr n. biol. Anita Gregor
Summary
The acute hepatic porphyrias are disorders that result from the inherited dysregulation of one of the enzymes in the porphyrin-heme biosynthetic pathway. The acute attack of porphyrias is a life threatening condition. Porphyrinogenic drugs, alcohol ingestion, reduced caloric intake due to fasting or dieting, infection and hormones might be precipitate these attacks. During attacks a variety of neurological and/or psychiatric symptoms may be occur which mimic many other disorders. Here we provide and overview of clinical, biochemical, genetical and other aspects of acute porphyrias that might be helpful providing more insight into these rare disorders.

Ostre porfirie wątrobowe są chorobami zależnymi od wrodzonych błędów metabolicznych biosyntezy hemu ujawniającymi się w klinicznej postaci pod wpływem działania różnorodnych czynników egzogennych i endogennych. Wyróżniamy cztery typy ostrych porfirii: ostrą przerywaną porfirię (AIP-Acute Intermittent Porphyria), porfirię mieszaną (VP – Variegate Porphyria), dziedziczną koproporfirię (HCP – Hereditary Coproporphyria) i porfirię z niedoboru ALA-dehydratazy (ALADP - ALA Dehydratase Deficiency Porphyria). Chociaż każdej ostrej porfirii odpowiada niedobór innego enzymu (ryc. 1), klinicznie są często trudne do rozróżnienia. Zespół objawów występujący w ostrych porfiriach może sugerować zupełnie inną jednostkę chorobową np.: ostre bóle brzucha (nasuwające podejrzenie zapalenia otrzewnej, porażenia jelit i pęcherza moczowego), porażenia mięśni doprowadzające niekiedy do niewydolności oddechowej, zaburzenia psychiczne sugerujące psychozę, a w części przypadków różnorodne zmiany skórne (11, 13, 18). Rozmaitość objawów prowadzi często do niewłaściwych rozpoznań i późniejszego leczenia lekami nasilającymi atak porfirii. Śmiertelność w pełnoobjawowym ataku choroby sięga około 10%. Wczesna, nawet niepełna diagnoza ma znaczenie rokownicze i pozwala uniknąć leczenia objawowego (często lekami szkodliwymi), niepotrzebnych laparotomii zwiadowczych prowadzących do rozwoju pełno objawowego ciężkiego napadu choroby. Porfiria jest chorobą często pomijaną w diagnostyce różnicowej bólów brzucha, chorób psychicznych czy też nietypowych polineuropatii. Porfirie, ze względu na różnorodną symptomatologię, nie należą do żadnej specjalności medycznej. Rzeczywista częstość występowania porfirii w Polsce nie jest znana. Na podstawie badań IHiT możemy w przybliżeniu określić, że 1 osoba obciążona porfirią przypada na około 10-15 tys. mieszkańców co daje w skali całego kraju około 4000-5000 osób (11). Na porfirię chorują głównie ludzie młodzi w wieku 20-40 lat. W materiale IHiT znajduje się 586 osób, które przebyły ataki ostrych porfirii wątrobowych.
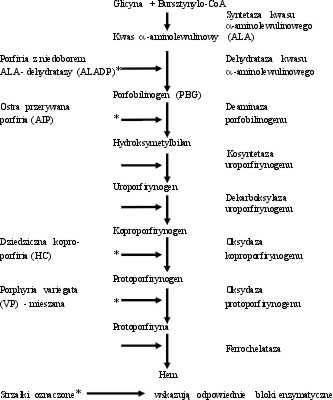
Ryc. 1. Schemat biosyntezy hemu i porfirie odpowiadające blokom enzymatycznym.
Rozpoznanie ostrej porfirii tylko na podstawie obrazu klinicznego jest niezmiernie trudne. Dlatego też w każdym przypadku musi być potwierdzone badaniami biochemicznymi i enzymatycznymi (tab. 1). Najbardziej typowo ostra porfiria wątrobowa (niezależnie od typu) objawia się w postaci napadów bólów brzucha i polineuropatii. Obraz kliniczny naśladuje rozmaite jednostki chorobowe, co utrudnia i opóźnia właściwe rozpoznanie pogarszając rokowanie. Początkowe, silne bóle brzucha i mięśni, zwłaszcza okolicy krzyżowej ustępują w miarę pojawiania się objawów neurologicznych: osłabienia, niedowładów mięśni i porażeń, także mięśni oddechowych. Pełnoobjawowemu atakowi towarzyszą objawy psychiczne: bezsenność, depresja, omamy, lęki. W porfirii mieszanej i w dziedzicznej koproporfirii u części chorych pojawiają się również zmiany skórne pod wpływem działania światła (5, 13). W pierwszym okresie bólów dochodzi często do laparotomii co wraz z podaniem leków do indukcji znieczulenia nasila objawy neurologiczne. Objawem najczęściej nasuwającym lekarzom podejrzenie porfirii jest czerwony mocz, ciemniejący po wystawieniu na światło słoneczne. Najgroźniejszym przebiegiem charakteryzuje się atak którego objawy narastają gwałtownie, w ciągu kilku godzin. Stosunkowo często występują u chorych objawy psychiczne w postaci zaburzeń snu, pobudzenia, agresji a nawet omamów (13).
Tabela 1. Rodzaj dziedziczenia i główne zmiany wykrywane badaniami biochemicznymi.
| Typ porfirii | Skrót nazwy | Rodzaj dziedziczenia | Główne zmiany biochemiczne |
| Ostra przerywana porfiria | AIP | Autosomalny dominujący | Mocz: ALAá, PBG |
| Porfiria mieszana | VP | Autosomalny dominujący | Mocz: PBG
mmmmporfiryny
Kał: protoporfiryny
mmmkoproporfiryny |
| Porfiria z niedoboru ALA-dehydratazy | ADP | Autosomalny recesywny | Mocz: ALA |
| Dziedziczna koproporfiria | HCP | Autosomalny dominujący | Mocz: koproporfiryny
Kał:mkoproporfiryny |
ALA – kwas d-aminolewulinowy, PBG – porfobilinogen.
Leczenie ataków i zaostrzeń jest podobne we wszystkich typach ostrych porfirii wątrobowych. W czasie zaostrzeń lub lekkich napadów bez nudności i wymiotów stosuje się glukozę doustnie (300-400 g/dobę), a w ciężkich stanach przez zgłębnik lub dożylnie w postaci 10% roztworu do żyły obwodowej, lub 20% roztworu glukozy do żyły centralnej. Glukoza hamuje, zwiększoną w czasie napadów, aktywność syntetazy kwasu delta-aminolewulinowego (ALA-S), enzymu regulującego szybkość przemiany porfiryn (2,11). W przypadkach ataku porfirii potwierdzonego dużym wydalaniem porfiryn i prekursorów hemu w moczu lekiem z wyboru jest Normosang. Argininian hemu (Normosang) stosuje się w dawce 3 do 4 mg/kg masy ciała na dobę we wlewie 100 ml 0,9% NaCl przez 2-4 kolejne dni. Ze względu na możliwość zapalenia żyły w miejscu wstrzyknięcia zaleca się każdorazowo odrębne wkłucia. Czas od przygotowania roztworu do zakończenia przetaczania nie powinien przekroczyć 30 min, gdyż powstające produkty degradacji mogą powodować objawy uboczne. Po przetoczeniu należy żyłę przepłukać solą fizjologiczną w obj. 200 ml. Poprawa kliniczna jest zazwyczaj widoczna po drugim przetoczeniu. Skuteczność leczenia napadu porfirii argininianem hemu maleje wraz z upływem czasu od wystąpienia pierwszych objawów. Wczesne zastosowanie leczenia glukozą, a zwłaszcza argininianem hemu powoduje ustąpienie objawów. Uzupełnienie niedoboru hemu prowadzi do zahamowania aktywności ALA-S. Inne leki podawane w celu łagodzenia objawów (leki przeciwbólowe, zwalniające częstość serca, uspokajające) mogą być stosowane o ile znajdują się na liście leków bezpiecznych. Każdy chory z ciężkim atakiem porfirii powinien być hospitalizowany, najlepiej w ośrodku posiadającym laboratorium pozwalające skutecznie różnicować porfirię. Algorytm postępowania w przypadku podejrzenia porfirii przedstawiono na rycinie 2. Rozpoznanie porfirii w okresie zaostrzenia choroby można ustalić badając wydalanie porfiryn i ich prekursorów, porfobilinogenu i kwasu γ-aminolewulinowego w moczu. Natomiast określenie poszczególnych typów ostrych porfirii możliwe jest po przeprowadzeniu badań enzymatycznych. Dzięki badaniom enzymatycznym możliwe jest wykrycie większości bezobjawowych nosicieli genu. Trudności sprawia wcale nie mała grupa nosicieli genu z graniczną lub prawidłową aktywnością poszczególnych enzymów. Tutaj rozstrzygające znaczenie mogą mieć badania genetyczne (4). Wiadomo, że u 80-90% nosicieli choroby nigdy nie dochodzi do ujawnienia się choroby.
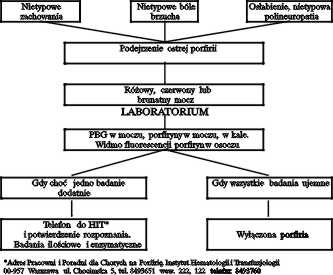
Ryc. 2. Algorytm postępowania w przypadku podejrzenia porfirii.
Obraz kliniczny różnych typów ostrych porfirii bywa często podobny (7). W wielu przypadkach postawienie prawidłowego rozpoznania typu porfirii może być bardzo trudne, szczególnie w przypadkach porfirii mieszanej. W VP często występują objawy neurologiczne i psychiczne podobne jak w AIP, HCP i porfirii z niedoboru ALA-D. Objawy niekoniecznie pojawiają się u wszystkich chorych jednakowo, ale występują z różnym natężeniem w poszczególnych przypadkach. Badania biochemiczne u bezobjawowych nosicieli genu AIP z granicznymi wartościami aktywności deaminazy porfirobilinogenu nie pozwalają na postawienie ostatecznego rozpoznania lub wykluczenia choroby. Rozwiązaniem mogłyby być w przyszłości badania wykrywające poszczególne mutacje genowe DNA (4, 10, 14). Pozwolą one być może na rzeczywiste i dokładne wykrycie wszystkich bezobjawowych nosicieli genu i bardzo precyzyjne określenie typu porfirii. W przypadku ostrych porfirii wątrobowych identyfikacja bezobjawowych członków rodzin ma zasadnicze znaczenie w profilaktyce zaostrzeń i ataków choroby. Podstawowym ograniczeniem badań genetycznych jest obecnie ich cena. W przyszłości analiza DNA będzie zapewne podstawowym badaniem różnicującym porfirie. Przyszłość to również ustalenie zależności pomiędzy poszczególnymi genotypami, a objawami klinicznymi i być może rozwój terapii genowej eliminującej błędy metaboliczne w porfirii (4, 12, 14).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Anderson K.E.: Semin Hematol 1989, 26:10.
2. Andreson Ch.: Umea and Arjeplog 1997.
3. Bonkowsky H., Schady W.: Sem Liver Dis 1982, 2:108.
4. Frank J., Christiano A.M.: Skin Pharmacol Appl Skin Physiol 1998; 11: 297.
5. Frank J., Christiano A.M.: Skin Pharmacol Appl Skin Physiol 1998, 11: 310.
6. Granick S.: J. Biol. Chem.: 1966, 241:1359.
7. Gregor A. et al.: of Ann. Med. 1994, 26, 125.
8. Kappas A. et al.: The porphyrias. In: Scriver CR Beudet AL, Sly WS, Valle D, eds. The Metabolic Basis of Inherited Disease. 6th ed. New York: McGraw-Hill, 1989.
9. Kauppinen R., Mustajoki P.: Medicine 1992, 71:1.
10. Kauppinen R.: National Public Health Institute, Helsinki, 1992.
11. Kostrzewska E. i wsp.: Porfirie (red. E. Kostrzewska i W. Kucharski) Instytut Hematologii i Transfuzjologii. Warszawa 1996.
12. Mazurier F. et al.: J Inherited Metab Dis 1997, 20: 247.
13. Moore M.R. et al.: Disorders of porphyrin metabolism. New York: Plenum Medical, 1987.
14. Petrides P.E.: Skin Pharmacol Appl Skin Physiol 1998, 11: 374
15. Tarczyńska-Nosol S.: Terapia i Leki 1999, XXVI/XLIX/1-2, 5.
16. Thunell S. et al.: J Stud Alcohol 1992, 53:272.
17. Tschudy D.P. et al.: Ann Intern Med 1975, 83:851.
18. Wilson J.H.P., de Rooij F.W.M.: Gastro-intestinal neurologic and psychiatric manifestations of acute porphyria. Porphyria Research Centre, University hospital Rotterdam-Dijk Zigt, Rotterdam, 1995.
