© Borgis - Postępy Nauk Medycznych 2/2001, s. 30-38
Marek Gacko
Tkankowy i osoczowy układ hemostatyczny w tętniaku aorty
Tissue and plasmatic haemostatic system in aortic aneurysm
Klinika Chirurgii Naczyń i Transplantacji Akademii Medycznej w Białymstoku
Kierownik Kliniki: prof. dr hab. Stanisław Głowiński
Streszczenie
W ścianie tętniaka aorty występuje urokinazowy aktywator plazminogenu, plazminogen i plazmina. Plazmina aktywuje prometaloproteazy. Degradacja białek strukturalnych przez plazminę i metaloproteazy przyczynia się do powiększania tętniaka. Lokalna aktywacja płytek krwi i osoczowych czynników krzepnięcia w poszerzeniu tętniakowym aorty prowadzi do powstania skrzepliny przyściennej. Gwałtowne i znaczne powiększenie tętniaka oraz zakażenie ściany tętniaka lub skrzepliny przyściennej może doprowadzić do uogólnionej aktywacji krzepnięcia i wystąpienia zespołu rozsianego wykrzepiania wewnątrznaczyniowego.
Summary
Urokinase plasminogen activator, plasminogen and plasmin are observed in the wall of aortic aneurysm. Plasmin activates prometalloproteases. Degradation of structural proteins by plasmin and metalloproteases contributes to enlargement of aortic aneurysm. Local activation of blood platelets and of plasmatic factors of coagulation in aneurysmal dilatation of aorta leads to formation of parietal thrombus. Rapid and marked enlargement of aneurysm and infection of the aneurysmal wall and parietal thrombus may result in general activation of coagulation and disseminated intravascular coagulation.

WSTĘP
Tętniak aorty stanowi wrzecionowate lub workowate rozszerzenie aorty o co najmniej 50% w porównaniu z odcinkiem niezmienionym. U osób powyżej 65 roku życia średnica aorty wynosi 2,01 ± 0,51 cm. Rozmiary tętniaków aorty osiągają średnicę od 3 do 15 cm. Tętniaki aorty występują u 2,4% populacji, pięciokrotnie częściej u mężczyzn niż u kobiet. Częstość występowania tętniaków w poszczególnych odcinkach aorty jest następująca: część wstępująca – 12,3%, łuk aorty – 8,0%, aorta piersiowa – 26,7%, aorta brzuszna powyżej odejścia tętnic nerkowych – 23,0%, aorta brzuszna poniżej odejścia tętnic nerkowych – 30,9% (41).
Powstanie tętniaka jest skutkiem uszkodzenia włókien elastycznych i utraty właściwości odwracalnego odkształcania ściany tętnic. Czynniki inicjujące zmiany doprowadzające do powstania tętniaka mogą być różne. Wiadomo natomiast, że degradacji elastyny i innych białek macierzy międzykomórkowej ściany aorty dokonują metaloproteazy i proteazy serynowe (55, 79, 82, 91), przy współudziale proteaz cysteinowych i proteaz aspartylowych (37).
W patologii tętniaka aorty znaczącą rolę odgrywają proteazy tkankowego oraz proteazy osoczowego układu krzepnięcia i fibrynolizy. W powstawaniu, powiększaniu i pękaniu tętniaka istotną rolę odgrywa urokinazowy aktywator plazminogenu i plazmina ściany aorty. Lokalna aktywacja płytek i osoczowego układu krzepnięcia doprowadza do powstania skrzepliny przyściennej wypełniającej światło tętniaka.
U chorych z tętniakiem aorty występuje często przewlekła, uogólniona, bezobjawowa aktywacja krzepnięcia krwi. Rzadziej pojawia się aktywacja objawowa w postaci zespołu rozsianego wykrzepiania wewnątrznaczyniowego, ze skazą krwotoczną i niewydolnością wielonarządową.
UDZIAŁ UROKINAZOWEGO AKTYWATORA PLAZMINOGENU I PLAZMINY W DEGRADACJI BIAŁEK STRUKTURALNYCH ŚCIANY TĘTNIAKA
Urokinazowy aktywator plazminogenu (uPA) syntetyzowany jest w postaci nieaktywnej, jako prourokinazowy aktywator plazminogenu (pro-uPA), w wielu rodzajach komórek. Prekursorem plazminy jest plazminogen (Plg). Plazminogen jest syntetyzowany w hepatocytach i granulocytach kwasochłonnych, skąd przechodzi do krwi i do przestrzeni międzykomórkowych. Pro-uPA wychwytywany jest przez receptory prourokinazowego aktywatora plazminogenu (pro-uPA/R), a plazminogen przez receptory plazminogenu (Plg/R) zlokalizowane na powierzchni błon komórkowych (3, 6). Powstające w wyniku autoaktywacji śladowe ilości plazminy dokonują aktywacji pro-uPA. Rozpoczyna to łańcuch reakcji aktywnych dalszych cząsteczek Plg przez urokinazę i aktywacji pro-uPA przez plazminę (ryc. 1). Związanie z receptorami znacznie zwiększa szybkość aktywacji pro-uPA i plazminogenu. Receptory te występują na powierzchni tych samych komórek, sąsiadują bezpośrednio ze sobą i spełniają rolę czynników zagęszczających (17). Związane z receptorem urokinaza i plazmina są mniej wrażliwe na działanie inhibitorów od enzymów wolnych i zachowują aktywność przez wiele godzin. Zachodzące z czasem zmiany konformacyjne cząsteczek uPA powodują, że do kompleksu uPA/R dołącza się inhibitor 1 aktywatora plazminogenu (PAI-1). Kompleks PAI-1/uPA/R wiąże się z białkiem pokrewnym receptora lipoprotein (lipoprotein receptor-related protein, LRP) (3, 5, 18). Związanie z LPR warunkuje internalizację kompleksu i przenoszenia go do lizosomów (10, 30, 56). W lizosomach uPA i PAI-1 jest degradowany i inaktywowany. Natomiast receptory uPA i LRP wracają na powierzchnię komórki i biorą udział w kolejnym cyklu aktywacji pro-uPA i eliminacji następnego nieaktywnego kompleksu PAI-1/uPA/R (ryc. 2). Identycznym przemianom podlega kompleks PI/R (31, 46, 73). Zapoczątkowuje je łączenie się tego kompleksu z a2-antyplazminą (a2-API). Zachodzące z udziałem LPR odtwarzanie receptora uPA i receptora Pl może zostać wyłączone przez białko towarzyszące receptorowi (receptor associated protein, RAP) (18, 64, 71). LPR związany z RAP nie wiąże PAI-1/uPA/R i a2-API/PI/R. Doprowadza to do obniżenia aktywności uPA i PI na powierzchni komórki.
Plazmina degraduje białka macierzy przestrzeni międzykomórkowych: fibronektynę, lamininę i inne glikoproteiny. Zapoczątkowuje ponadto aktywację wielu prometaloproteaz (9, 21, 59, 68). Wśród aktywowanych MMPs są elastazy, kolagenazy, żelatynazy i stromelizyny (proteoglikanazy). Aktywność MMPs jest regulowana przez tkankowe inhibitory metaloproteaz (tissue inhibitors metalloproteases, TIMPs).

Ryc. 1. Ciągła aktywacja prourokinazowego aktywatora plazminogenu (pro-uPA) do urokinazowego aktywatora plazminogenu (uPA) i plazminogenu (Plg) do plazminy (PI) zachodząca na powierzchni komórki.
pro-uPA/R receptor prourokinazowego (urokinazowego) aktywatora plazminogenu, Plg/R - receptor.
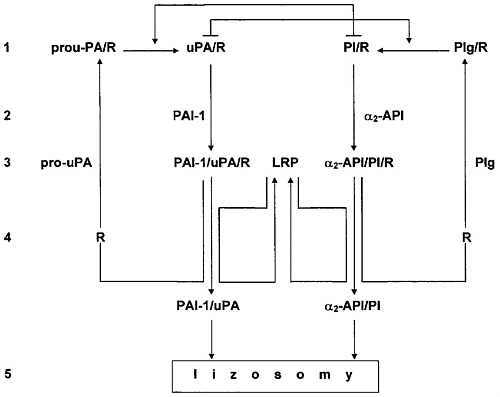
Ryc. 2. Udział receptorów zagęszczających (R) i receptora klarującego (LRP) w cyklu aktywacji prourokinazowego aktywatora plazminogenu (pro-uPA) i plazminogenu (Plg).
Objaśnienia: ProuPA(uPA)/R - receptor prourokinazowego (urolinazowego aktywatora plazminogenu, Plg/R (PI/R) - receptor plazminogenu (plazminy), PAI - 1 - inhibitor 1 aktywatora plazminogenu, a2-API - alfa2-antyplazmina, PAI-1/uPA/R - kompleks inhibitor 1 aktywatora plazminogenu/urokinazowy aktywator plazminogenu/receptor, PAI-1/uPA - kompleks inhibitor 1 aktywatora plazminogenu/urokinazowy aktywator plazminogenu, a2-API/PI/R - kompleks alfa2-antyplazmina/plazmina/receptor, a2-API/PI - kompleks alfa2-antyplazmina/plazmina.
1 - aktywacja proenzymów, 2 - hamowanie enzymów, 3 - internalizacja kompleksu inhibitor-enzym-receptor, 4 - recyklizacja receptora, 5 - degradacja inhibitora i enzymu w lizosomach.
W ścianie tętniaka aorty wykazano zwiększoną aktywność urokinazowego aktywatora plazminogenu i zwiększoną aktywność plazminy w porównaniu do ściany aorty prawidłowej (51, 75, 81). Szczególnie wysoką aktywnością tych proteaz odznaczają się powstające w ścianie tętniaka vasa vasorum (47, 49). Równocześnie obserwowano obniżenie aktywności PAI-1 (76, 85, 92). W ścianie tętniaka wykazano również zwiększoną aktywność wielu metaloproteaz i obniżoną zawartość ich inhibitorów (19, 30, 35, 69, 70, 89, 96). Wskazuje to na przesunięcie równowagi proteolityczno-antyproteolitycznej na korzyść plazminy i MMPs oraz ujawnienie się ich aktywności. Degradacja białek strukturalnych przez plazminę i metaloproteazy powoduje lokalne obniżenie wytrzymałości mechanicznej ściany tętnicy (ryc. 3).
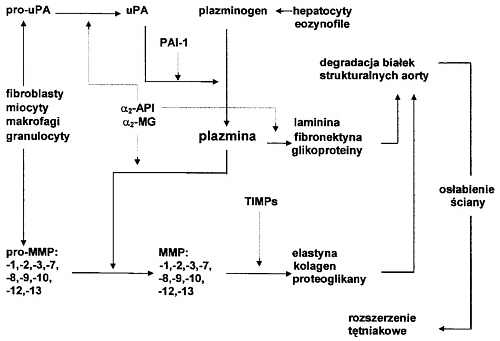
Ryc. 3. Rola plazminy i metaloproteaz w degradacji białek strukturalnych aorty i w powstawaniu tętniaka.
Objaśnienia: pro-MMP (MMP) - prometaloproteazy (metaloproteazy), TIMPs - tkankowe inhibitory metaloproteaz. Pro-uPA (uPA) prourokinazowy (urokinazowy) aktywator plazminogenu, PAI-1 - inhibitor 1 aktywatora plazminogenu, a2-API - alfa2-antyplazmina, a2-MG - alfa2-makroglobulina, proMMP/MMP - prometaloproteazy (metaloproteazy), TIMPs - tkankowe inhibitory metaloproteaz. Linia: ciągła - uwalnianie, aktywacja, degradacja; przerywana - hamowanie.
W celu zahamowania degradacji białek strukturalnych i zwolnienia powiększania się tętniaka podejmowane są próby podawania inhibitorów proteaz o różnym mechanizmie działania i różnej specyficzności. Należy do nich kwas e-aminokapronowy, kwas traneksamowy, traskolan, marimastat, deoksycyklina, kaptopryl i enalapryl (4, 13, 23, 34,90).
ZABURZENIA HEMOSTAZY U CHORYCH Z TĘTNIAKIEM AORTY
Aktywacja płytek i czynników krzepnięcia krwi u chorych z tętniakiem aorty może być ograniczona do miejsca tętniakowego rozszerzenia aorty i zachodzić na powierzchni uszkodzonych komórek lub na powierzchni włókien kolagenowych (w fazie stałej). Może także mieć charakter uogólniony, obejmować krew krążącą i dokonywać się w fazie płynnej osocza. Płytki krwi wiążą się, za pośrednictwem czynnika von Willebranda, z kolagenem odsłoniętej w stanach patologicznych warstwy podśródbłonkowej. Prowadzi to do powstania zlepu płytkowego. Aktywacja czynników krzepnięcia w fazie stałej, ze względu na ich zagęszczenie, bezpośrednie wzajemne pobliże i korzystny układ przestrzenny jest 104-105 razy szybsza niż w osoczu. Związane z powierzchnią komórki lub włóknami kolagenu czynniki są ponadto niedostępne dla inhibitorów osoczowych. W fazie stałej zachodzą cztery kluczowe etapy aktywacji krzepnięcia krwi. Na powierzchni uszkodzonych komórek tworzy się kompleks TF-VII-Ca2+ warunkujący powstanie aktywnego czynnika VII. Szybkiej aktywacji czynnika XII i prekalikreiny sprzyja związanie się tych proenzymów i wielkocząsteczkowego kininogenu z włóknami kolagenu warstwy podśródbłonkowej naczyń lub z powierzchnią aktywnych płytek krwi. Na fosfolipidach powierzchni błon komórkowych płytek powstaje kompleks IXa-VIIIa-X-Ca2+, w którym aktywowany jest czynnik X. Z udziałem fosfolipidów błonowych tworzy się także kompleks Xa-Va-II-Ca2+ warunkujący powstanie trombiny.
Lokalna i uogólniona aktywacja układu hemostatycznego występująca w przebiegu tętniaka aorty może przebiegać w sposób utajony lub manifestować się objawami klinicznymi (tab. 1). W przypadku lokalnej utajonej aktywacji krzepnięcia badania laboratoryjne krwi pobranej w miejscu odległym nie wykazują zmian lub wykazują jedynie niewielkie zmiany. W lokalnej objawowej nadkrzepliwości występującej bardzo często w tętniaku aorty obserwuje się zmiany koagulacyjne we krwi krążącej dotyczące jedynie wczesnej fazy aktywacji krzepnięcia krwi, takie jak pojawienie się zwiększonego stężenia peptydu uwalnianego w czasie aktywacji czynnika IX i X oraz fragmentu 1+2 protrombiny. Nadkrzepliwość uogólniona, utajona przebiega z obniżeniem stężenia antytrombiny III, zwiększeniem stężenia kompleksów trombina-antytrombina III (97) ale z niecharakterystycznymi objawami klinicznymi (83, 84). U 0,5-1,0% chorych z tętniakiem aorty występuje nadkrzepliwość uogólniona objawowa określana jako rozsiane wykrzepianie wewnątrznaczyniowe (disseminated intravascular coagulation, DIC) (1, 54, 56). Charakteryzuje się ono powstaniem agregatów płytkowych i złogów fibryny w mikrokrążeniu z klinicznymi konsekwencjami tych zaburzeń.
Tabela 1. Podział nadkrzepliwości występujących w przebiegu tętniaka aorty.
| Nadkrzepliwosc | Utajona | Objawowa |
| Lokalna | zakrzepy przyscienne |
| przejsciowe | dlugotrwale |
| zaburzenia koagulacyjne |
| brak lub niewielkie | niewielkie |
| objawy kliniczne |
| brak | objawy zakrzepu tetniczego |
| Uogólniona | mikrozakrzepy |
| przejsciowe | dlugotrwale |
| wybroczyny |
| nie wystepuja | wystepuja |
| zaburzenia koagulacyjne |
| znaczne | charakterystyczne dla DIC |
| objawy kliniczne |
| niecharakterystyczne | wstrzas, niewydolnosc nerek |
Ograniczenie aktywacji krzepnięcia krwi do miejsca tętniakowego rozszerzenia aorty lub uogólnienie aktywacji krzepnięcia zależy od wielu czynników: wielkości powierzchni uszkodzenia śródbłonka, dołączenia się zakażenia bakteryjnego, wirusowego lub grzybiczego oraz sprawności mechanizmów wyrównawczych i systemu obronnego ustroju. Zaktywowane czynniki krzepnięcia są rozcieńczane przez przepływającą krew i inaktywowane przez inhibitory. Znaczną rolę w zapobieganiu uogólnionemu krzepnięciu śródnaczyniowemu spełnia układ fagocytów jednojądrzastych wątroby, śledziony i płuc. Wychwytują one uszkodzone płytki krwi, aktywne czynniki krzepnięcia i monomery fibryny, które są degradowane w lizosomach (74). Liczba lizosomów i aktywność występujących w nich proteaz w czasie fagocytozy zwiększa się. Po rozległych operacjach i urazach oraz w zakażeniach bakteryjnych następuje wyczerpanie układu fagocytów jednojądrzastych. Istotne znaczenie posiada stan funkcjonalny wątroby, w której syntetyzowane są nie tylko czynniki krzepnięcia rodziny protrombiny, ale także inhibitory krzepnięcia: antytrombina III, kofaktor II heparyny oraz białko C i białko S.
Uszkodzony w miejscu rozszerzenia tętniakowego śródbłonek traci właściwości antykoagulacyjne i nabywa właściwości prozakrzepowe. Odsłonięta warstwa podśródbłonkowa wykazuje znaczną aktywność prokoagulacyjną. Decyduje o niej wysoka zawartość czynnika tkankowego i bardzo niska zawartość aktywatorów plazminogenu oraz odsłonięcie włókien kolagenu, które aktywują czynnik XII i stanowią miejsce adhezji płytek (25, 72). Istotne znaczenie w aktywacji krzepnięcia posiada także zmiana kształtu aorty i warunków przepływu krwi w obrębie rozszerzenia tętniakowego, na które składa się zwolnienie prądu krwi, zawirowania i zalegania krwi oraz zmiana przepływu laminarnego krwi w turbulentny (95). Aktywacji krzepnięcia sprzyja wzrost lepkości krwi.
W większości przypadków o powstaniu zakrzepu lub wystąpieniu krwawienia decydują zaburzenia równowagi stosunku aktywatorów protrombiny i aktywatorów plazminogenu do inhibitorów tych aktywatorów oraz stosunku protrombiny i plazminogenu do inhibitorów trombiny i plazminy (ryc. 4). Istnieją jednak sytuacje kliniczne niemieszczące się w przedłożonym schemacie. U chorych z ostrą lub podostrą postacią rozsianego wykrzepiania wewnątrznaczyniowego obserwuje się rozsiane skrzepliny w mikrokrążeniu z równoczesną skazą krwotoczną powstałą w wyniku zużycia czynników krzepnięcia.

Ryc. 4. Zakrzep lub krwotok jako konsekwencja zaburzeń równowagi układu aktywacji protrombiny i plazminogenu.
PRZYŚCIENNA AKTYWACJA KRZEPNIĘCIA KRWI
W tworzeniu zakrzepu w naczyniach tętniczych główną rolę odgrywają płytki krwi. Ze strony aorty miejsce kontaktu z płytkami stanowią pozbawione śródbłonka rozszerzenia tętniakowe i zmiany miażdżycowe. W warunkach szybkiego przepływu krwi w tętnicach i wysokiego napięcia ścinającego ulegają odsłonięciu receptory błonowe płytek warunkujące ich adhezję (77, 78). Stanowi je kompleks glikoprotein Ib-V-IX wiążący czynnik von Willebranda, który pośredniczy w adhezji płytek do włókien kolagenu. Zainicjowane równocześnie zmiany cytoszkieletu płytki powodują uwalnianie do środowiska ich zawartości, w tym ADP i tromboksanu A2. Związki te odsłaniają receptory IIb-IIIa uczestniczące w agregacji płytek. W tym samym czasie następuje aktywacja zewnątrzpochodnego i wewnątrzpochodnego układu krzepnięcia krwi oraz przyspieszenie aktywacji czynnika IX przez aktywny czynnik VII (20). Powstające włókna fibryny umacniają czop płytkowy. Związana z fibryną trombina jest niewrażliwa na działanie AT-III. Trombina jest silnym czynnikiem agregującym. Enzym ten odsłania fosfolipidy na powierzchni płytek oraz aktywuje czynnik V i czynnik VIII. Nieco później dochodzi do uwolnienia tkankowego aktywatora plazminogenu (tPA) i aktywacji fibrynolizy. Dzięki temu powstająca plazmina działa na utworzoną już fibrynę (ryc. 5).
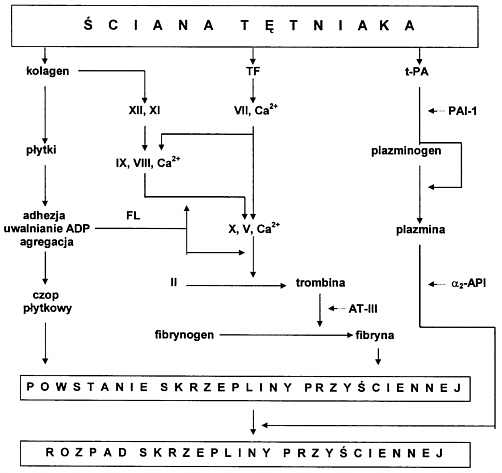
Ryc. 5. Powstawanie i rozpad skrzepliny przyściennej wypełniającej światło tętniaka.
Objaśnienia: AT-III - antytrombina III, a2-API - alfa2-antyplazmina, FL - fosfolipidy płytek krwi, PAI-1 - inhibitor 1 aktywatora plazminogenu, TF - czynnik tkankowy, t-PA - tkankowy aktywator plazminogenu.
W wyniku lokalnej aktywacji krzepnięcia krwi tętniak aorty wypełnia z reguły skrzeplina przyścienna (86). Jej obecność można potwierdzić śledząc gromadzenie się znakowanych 111In-płytek (53) lub znakowanego 125I-fibrynogenu (22). Jak wynika z tabeli 2 skrzeplina przyścienna wykazuje znaczną aktywność czynnika tkankowego i znaczną aktywność antyheparynową. Są one wyższe niż w skrzepie krwi. Skrzeplina nie wykazuje natomiast aktywności antytrombinowej. Skrzeplina przyścienna wykazuje także znacznie wyższą aktywność aktywatorów plazminogenu niż skrzep krwi i zawiera około pięciokrotnie więcej plazminogenu. Skrzeplina przyścienna nie zawiera natomiast antyplazmin. Brak antytrombin i antyplazmin w skrzeplinie przyściennej może być spowodowany inaktywacją tych inhibitorów przez leukocytarną elastazę (16, 27, 52). Enzym ten inaktywuje także inhibitory aktywatorów plazminogenu (93). Wysoka aktywność czynnika tkankowego i czynników neutralizujących antykoagulacyjne działanie heparyny oraz brak substancji działających antytrombinowo sprzyja powiększaniu się skrzepliny przyściennej wypełniającej światło tętniaka. Narastanie skrzepliny ogranicza jednak wysoka aktywność aktywatorów plazminogenu, znaczna zawartość plazminogenu i brak antyplazmin. Dzięki równowadze koagulacyjno-fibrynolitycznej skrzeplina wypełniająca rozszerzenie tętniakowe nie powiększa się nadmiernie, nie zmniejsza przepływu krwi i nie doprowadza do niedrożności aorty. Po wypełnieniu poszerzenia tętniakowego powierzchnia skrzepliny kontaktuje się z szybkim prądem krwi, a powstające na jej powierzchni aktywne czynniki krzepnięcia i monomery fibryny są rozcieńczane i inaktywowane przez inhibitory osoczowe.
Tabela 2. Aktywność składników układu krzepnięcia i fibrynolizy skrzepliny przyściennej tętniaka aorty i obkurczonego skrzepu krwi (38), z uzupełnieniem.
| Aktywnosc | Skrzep krwi | Skrzeplina przyscienna tetniaka |
| Czynnik tkankowy, s | 89,0 ? 10,2 | 29,0 ? 7,4* |
| Aktywnosc antytrombinowa, s | 15,0 ? 0,1 | 16,0 ? 1,1 |
| Aktywnosc antyheparynowa, s | 24,8 ? 1,7 | 16,2 ? 1,6* |
| Aktywatory plazminogenu, j/ml** | 122,0 ? 19,2 | 160,1 ? 32,5* |
| Plazminogen, Tyr, nmol/ml | 50,7 ? 3,2 | 256,1 ? 45,9* |
| Antyplazminy, j/ml*** | 8,9 ? 0,3 | 0,0 |
Różnica istotna statystycznie:
*** p < 0,05.
*** 1 jednostkę aktywności aktywatorów plazminogenu stanowi skrócenie czasu fibrynolizy o 1 minutę w porównaniu do kontroli.
*** 1 jednostkę aktywności antyplazminy stanowi różnica ilości nmol/ml uwolnionej tyrozyny w próbie kontrolnej i badanej.
Skrzeplina przyścienna obniża działanie sił hemodynamicznych na ścianę tętniaka oraz poprawia warunki przepływu krwi przez rozszerzenie tętniakowe i może w ten sposób zwalniać jego powiększanie (65). Pogarsza jedynie odżywianie ściany tętniaka, a odrywanie się fragmentów skrzepliny może doprowadzić do zatorów tętnic obwodowych (3).
UOGÓLNIONA AKTYWACJA KRZEPNIĘCIA KRWI
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Aboulafia D.M., Aboulafia E.D.: Ann. Vasc. Surg. 1996, 10:396.
2. Adam D.J. et al.: J. Vasc. Surg. 1999, 30:641.
3. Adolph R. et al.: J. Vasc. Surg. 1997, 25:916.
4. Allaire E. et al.: Circulation,1998, 98:249.
5. Andreasen P. et al.: FEBS Lett. 1994, 338:239.
6. Agraves K. et al.: J. Biol. Chem. 1995, 270:26550.
7. Baglin T.: Brit. Med. J.,1996, 312:683.
8. Balduini C.L. et al.: Haematologica 1997, 82:581.
9. Baramova E.M. et al.: FEBS Lett. 1997, 405:157.
10. Bick R.L., Kunkel L.A.: Int. J. Haematol. 1992, 55:1.
11. Booth N.A. et al.: Clin. Lab. Haematol. 1984, 64:123.
12. Bowald S., Gerdin B.: Acta Chir. Scand. 1980, 146:351.
13. Boyle J. et al.: J. Vasc. Surg. 1998, 27:354.
14. Bradbury A.W. et al.: J. Vasc. Surg. 1995, 21:481.
15. Brothers T.E. et al.: J. Inveset. Surg. 1993, 6:527.
16. Brower M.S., Harpel P.C.: J. Biol. Chem. 1982, 257:9849.
17. Bu G. et al.: Blood 1994, 83:3427.
18. Bu G. et al.: Proc. Nat. Acad. Sci. 1992, 89:7427.
19. Busuttil R.W., Abou-Zamzam A.M., Machleder H.I.: Arch. Surg. 1980, 115:1373.
20. Camerer E. et al.: Thrombos. Res. 1996, 81:1.
21. Carmeliet P. et al.: Nat. Genet. 1997, 17:439.
22. Cate J.W. et al.: Am. J. Med. 1975, 59:171.
23. Cawston T.E.: Pharmacol. Ther. 1996, 70:163.
24. Chauvet V. et al.: Ann. Fr. Anesth. Reanim. 1991, 10:164.
25. Cocheri S., Astrup T.: Proc. Soc. Exp. Biol. Med. 1961, 108:369.
26. Cohen J.R. et al.: Ann. Vasc. Surg. 1987, 1:552.
27. Cohen J.R. et al.: Am. J. Surg. 1990, 56:665.
28. Coto H. et al.: J. Cardiovasc. Surg. 1985, 26:280.
29. Davies M.J. et al.: Br. J. Surg. 1993, 80:974.
30. Dobrin P.B. et al.: Arch. Surg. 1984, 119:405.
31. Felez J.: Fibrinolysis Proteolysis 1998, 12:183.
32. Fisher D.F. et al.: Arch. Surg. 1993, 118:1252.
33. Fouser L.S. et al.: Am. J. Clin. Pathol. 1980, 74:701.
34. Franklin I.J. et al.: Br. J. Surg. 1999, 86:771.
35. Freestone T. et al.: Arterioscler. Thromb. Vasc. Biol. 1995, 15:1145.
36. Gaciong Z. et al.: Acta Angiol. 1998, 4, supl. 1:16.
37. Gacko M., Głowiński S.: Clin. Chem. Lab. Med. 1998, 36:449.
38. Gacko M. et al.: Ann. Acad. Med. Bialost. 1999, 44:102.
39. Gertler J.P. et al.: J. Vasc. Surg. 1996, 24:936.
40. Getaz E.P., Louw J.H.: Postgrad Med. J. 1977, 53:668.
41. Gloviczki P.: J. Vasc. Surg. 1990, 11:19.
42. Glowiński S. et al.: Ann. Acad. Med. Bialost. 1995, 40:156.
43. Głowiński S. et al.: Polimery Med. 1992, 22:31.
44. Głowinski S. et al.: Polimery Med. 1992, 22:17.
45. Goto H. et al.: J. Vasc. Surg. 1985, 26:280.
46. Hajjar K.: Thrombos. Haemostas. 1995, 74:294.
47. Herron G.S. et al.: Arteriosclerosis Thromb. Vasc. Biol. 1991, 11:1667.
48. Holmberg A. et al.: Thrombos. Res. 1999, 96:99.
49. Holmes D.R. et al.: J. Vasc. Surg. 1995, 21:761.
50. Ikeda U., Shimada K.: Amer. J. Surg. 1999, 177:527.
51. Jean-Claude J. et al.: Surgery 1994, 116:472.
52. Jordan R.E. et al.: J. Biol. Chem. 1989, 264:10493.
53. Kanda T. et al.: Angiology 1993, 44:420.
54. Kazmers A. et al.: J. Vasc. Surg. 1996, 23:191.
55. Lee A.J. et al.: Blood Coagulat. Fibynol. 1996, 6:695.
56. Levi M., Cate H.T.: New Engl. J. Med. 1999, 341:586.
57. Lim R.C. et al.: Surg. Forum. 1967, 18:25.
58. Malanowicz W., Skarżyńska M.: Pol. Arch. Med. Wewn. 1975, 54:543.
59. Mazzieri R. et al.: EMBO J. 1997, 16:2319.
60. Mc Kay D.G.W.: Thrombos. Diathes. Haemorrhag. 1969, supl., 26:67.
61. Meissner M.H. et al.: Vasc. Surg. 1997, 31:727.
162. Micallef-Eynaud P.D., Ludlam C.A.: Blood Coagulat. Fibrinol. 1991, 2:477.
163. Milne A.A. et al.: Eur. J. Vascular. Surg. 1994, 8:622.
164. Moestrup S.K., Gliemann J.: Biol. Chem. 1991, 266:14011.
165. Mower W.R. et al.: J. Vasc. Surg. 1997, 26:602.
166. Mulcare R.J. et al.: Surg. Gynecol. Obstet. 1976, 143:730.
167. Mulcare R.J. et al.: Ann. Surg. 1974, 180:343.
168. Murphy G.S. et al.; Ann. NY Acad. Sci. 1992, 667:1.
169. Newman K.M. et al.: J. Vasc. Surg. 1994, 20:814.
170. Newman K.M. et al.: Atheroscler. Thrombos. 1994, 14:1315.
171. Nielsen M.S. et al.: J. Biol. Chem. 1995, 270:23713.
172. Omoyama K., Takeda K.: Thrombos. Diathes. Haemorrhag. 1969, 21:1.
173. Plow E.F. et al.: Thrombos. Haemostas. 1991, 66:32.
174. Prose P.H.: Am. J. Pathol. 1965, 47:403.
175. Reilly J.M.: Ann. NY Acad. Sci. 1996, 800:151.
176. Reilly J.M., Sicard G.A., Lucore C.L.: J. Vasc. Surg. 1994, 19:865.
177. Roth G.J.: w: Molecular basis of thrombosis and haemostasis, Marcel Dekker, Inc., New York, Basel, Hong Kong 1995, 561.
178. Ruggeri Z.M.: Thrombos. Haemostas. 1993, 70:119.
179. Sakai N. et al.: Neurosurgery 1999, 45:34.
180. Satta J. et al.: J. Vasc. Surg. 1996, 737.
181. Schneiderman J. et al.: Am. J. Pathol. 1998, 152:703.
182. Schneiderman J. et al.: J. Clin. Invest. 1995, 96:639.
183. Schnetzer G.W., Penner J.A.: South Med. J. 1973, 66:264.
184. Siebert W.T., Natelson E.A.: Arch. Surg. 1976, 11:539.
185. Shireman P.K. et al.: J. Vasc. Surg. 1997, 25:157.
186. Stehbens W.E.: Thrombos. Haemostas. 1997, 78:952.
187. Sułek K.: Acta Haematol. Pol. 1997, 28, suppl. 2:148.
188. Szczawiński W. i wsp.: Lek. Wojsk. 1999, 3-4:213.
189. Thompson R.W. et al.: J. Clin. Invest. 1995, 96:318.
190. Threharne G.D. et al.: Br. J. Surg. 1999, 86:1053.
191. Tilson M.D., Newman K.M.: w: Aneurysms. New Findings and Treatments, red. J.S.T. Yao, W.H. Pearse. C.T.: Apleeton and Lange Norwalk, 1994, 3.
192. Tromholt N. et al.: Eur. J. Vasc. Surg. 1993, 7:675.
193. Urano T. et al.: Pol. J. Pharmacol. 1996, 48:209.
194. Yamazumi K. et al.: Am. J. Surg. 1998, 175:297.
195. Yu S.C.M., Zhao J.B.: Med. Eng. Phys. 1999, 21:133.
196. Zarins C.K. et al.: J. Vasc. Surg. 1986, 3:238.
197. Zawilska K.: Acta Haematol. Pol. 1994, 25, suppl. 2:27.
198. Zawilska K.: Acta Haematol. Pol. 1995, 26:33.
199. Zawilska K.: Nowiny Lek. 1992, 4:14.
100. Zawilska K.: Acta Haematol. Pol. 1999, 30, suppl. 1:203.
