© Borgis - Postępy Nauk Medycznych 4/2004, s. 4-11
Stanisław Czekalski1, Andrzej Ciechanowicz2
Mechanizmy i znaczenie sodowrażliwości ciśnienia tętniczego*
Mechanisms and importance of sodium sensitivity of blood pressure
1z Katedry i Kliniki Nefrologii, Transplantologii i Chorób Wewnętrznych Akademii Medycznej w Poznaniu
Kierownik Kliniki: prof. dr hab. med. Stanisław Czekalski
2z Samodzielnej Pracowni Patobiochemii i Biologii Molekularnej Pomorskiej Akademii Medycznej w Szczecinie
Kierownik Pracowni: dr hab. med. Andrzej Ciechanowicz
Streszczenie
Fenotypowa cecha sodowrażliwości ciśnienia tętniczego jest cechą jakościową o różnej ekspresji u ludzi uwarunkowanej w znacznym stopniu ilością sodu dostarczonego do organizmu. Uzyskano dowody, że sodowrażliwość ciśnienia tętniczego jest związana z działaniem czynników, które przewlekle nasilają resorpcję sodu w cewkach nerkowych, zarówno dystalnych (monogenowe postacie nadciśnienia) jak i proksymalnych. Cecha sodowrażliwości może być uwarunkowana genetycznie, najprawdopodobniej wielogenowo. Może być także cechą nabytą wywołaną subtelnym uszkodzeniem nerek oraz wywołaną otyłością i występującą w nadciśnieniu wtórnym. Przedstawiono hipotezę, że sodowrażliwość ciśnienia może być często związana z wrodzoną zmniejszoną liczbą nefronów spowodowaną działaniem czynników genetycznych i/lub środowiskowych w życiu płodowym. Występowanie cechy sodowrażliwości ciśnienia tętniczego jest czynnikiem ryzyka rozwoju nadciśnienia i zwiększonej śmiertelności.
Summary
Phenotypic trait of sodium sensitivity of blood pressure is the qualitative trait with different expression in humans, determined in large extent by the quantity of sodium delivered to organism. There are proves that blood pressure sodium sensitivity is associated with the action of factors which increases chronically sodium reabsorbtion in renal tubules both distal (monogenic forms of hypertension) and proximal. The trait of sodium sensitivity may be genetically determined, most probably by multiple genes. It may be also acquired, caused by the subtle renal injury, obesity, and is present in secondary forms of arterial hypertension. The hypothesis was proposed that sodium sensitivity of blood pressure may be often associated with congenital reduced nephron number caused by genetic and/or environmental factors acting in fetal life. Sodium sensitivity of blood pressure is a risk factor for the development of arterial hypertension and increased mortality.

Ciśnienie tętnicze jest sodowrażliwe gdy ulega podwyższeniu pod wpływem zwiększonego dostarczenia sodu do organizmu drogą doustną lub parentalną, natomiast obniża się przy ograniczeniu dostarczania sodu. Jeżeli ciśnienie tętnicze nie zmienia się pod wpływem różnych ilości dostarczanego do organizmu sodu, lub nawet obniża się przy zwiększaniu dostarczonego sodu, wówczas ciśnienie tętnicze określa się jako sodoniewrażliwe lub rzadziej jako sodooporne.
Fenotypowa cecha sodowrażliwości ciśnienia tętniczego jest cechą jakościową o różnej ekspresji u ludzi, uwarunkowanej w znacznym stopniu ilością sodu dostarczonego do organizmu. Już w 1979 roku Luft i wsp. (1) wykazali, że zwiększając progresywnie, co trzy dni, ilość spożywanego sodu od 10 mmol/dobę do 300, 600-800 i 1200-1500 mmol/dobę można było wykazać podwyższenie średniego ciśnienia tętniczego (mean arterial pressure, MAP) u wszystkich 16 badanych młodych, zdrowych, normotensyjnych osób. Podwyższenie wartości MAP było jednak bardzo różne u poszczególnych osób i przy największym spożywaniu sodu mieściło się w szerokich granicach od 1,5-34% wartości wyjściowej, zarejestrowanej przy spożywaniu 10 mmol sodu na dobę. Wyniki tego badania przekonują, że przy bardzo dużej ilości sodu dostarczonego do organizmu u wszystkich ludzi dochodzi do podwyższenia ciśnienia tętniczego. Świadczy to, że pomimo uruchomienia wszystkich złożonych mechanizmów zapobiegających zmianom środowiska wewnętrznego organizmu, przy bardzo różnym w indywidualnych przypadkach dostarczeniu sodu do organizmu, niezbędne staje się podwyższenie ciśnienia tętniczego dla utrzymania bilansu sodowego, czyli zrównoważenia podaży sodu i jego wydalania przez nerki. Przyczyn ujawnienia się cechy sodowrażliwości ciśnienia przy mniejszej lub większej podaży sodu u poszczególnych osób upatruje się w różnej sprawności mechanizmów odpowiedzialnych za zjawisko natriurezy ciśnieniowej. Złożone mechanizmy współdziałające w zjawisku natriurezy ciśnieniowej zostały tylko częściowo poznane, a ich dokładniejsze omówienie zawierają inne publikacje (2, 3). Fizjologiczna sprawność mechanizmów natriurezy ciśnieniowej może ulegać zaburzeniu pod wpływem różnych czynników hemodynamicznych, neurohumoralnych i hormonalnych. Gdy jakiś czynnik zakłóca przewlekle mechanizm natriurezy ciśnieniowej, to dla utrzymania równowagi gospodarki sodowej organizmu dochodzi do podwyższenia ciśnienia tętniczego wskutek hiperwolemii i (lub) wazokonstrykcji. Podwyższenie ciśnienia tętniczego umożliwia skompensowanie wpływu czynnika zaburzającego mechanizm natriurezy ciśnieniowej i ustalenie nowego stanu równowagi między ilością sodu dostarczonego do organizmu i jego wydalaniem przez nerki.
Badania doświadczalne dostarczyły dowodów, że cecha sodowrażliwości ciśnienia tętniczego jest związana z działaniem czynników, które przewlekle nasilają reabsorpcję sodu w cewkach nerkowych. U ludzi trzy rzadkie postacie nadciśnienia tętniczego uwarunkowanego mutacją jednego genu związane są z nasiloną reabsorpcją sodu przez nerki zlokalizowaną w cewkach dalszych nefronów (4). Zespół Liddla spowodowany jest skróceniem lub mutacją karboksyterminalnych odcinków podjednostek b lub g nabłonkowego kanału sodowego wrażliwego na amilorid. Mutacje te powodują zwiększony przezbłonowy transport sodu i stałą nasiloną jego reabsorpcję w cewce dalszej nefronu. Skutkiem jest wczesny rozwój sodowrażliwego nadciśnienia tętniczego z hipokaliemią, niską aktywnością reninową osocza i niskimi stężeniami aldosteronu w osoczu. Korekta tego zaburzenia przez zastosowanie triamterenu i amiloridu w połączeniu z dietą niskosodową powoduje normalizację ciśnienia tętniczego i ustąpienie towarzyszących mu nieprawidłowości. Hiperaldosteronizm poddający się leczeniu glukokortykoidami (GRA) jest spowodowany występowaniem chimerycznego genu 11 beta-hydroksylazy/syntazy aldosteronowej, co powoduje stałą zwiększoną syntezę aldosteronu w korze nadnerczy prowadzącą do nasilonej dystalnej reabsorpcji sodu w nefronach i rozwój sodowrażliwego nadciśnienia tętniczego. Zahamowanie zwiększonej syntezy aldosteronu przez leczenie glukokortykoidami powoduje normalizację ciśnienia tętniczego. Pozorny nadmiar mineralokortykoidów (AME) powstaje w wyniku mutacji genu nerkowospecyficznej izoformy 11-beta-dehydrogenazy hydroksysteroidowej, co powoduje, że białko będące produktem zmutowanego genu jest niezdolne do przekształcenia kortyzolu w kortykosteron. W dystalnej cewce nefronu przekształcenie to jest niezbędne w celu ochrony receptorów dla mineralokortykoidów, które mają takie same powinowactwo do kortyzolu jak do aldosteronu. Niechronione receptory dla mineralokortykoidów wiążą kortyzol, co powoduje nasiloną reabsorpcję sodu w cewkach dystalnych nefronów i rozwój sodowrażliwego nadciśnienia tętniczego, poddającego się leczeniu za pomocą tiazydowych diuretyków i spironolaktonu.
Scharakteryzowane powyżej jednostki chorobowe dostarczyły dowodów, że u ludzi uwarunkowana genetycznie nasilona reabsorpcja sodu w dystalnym odcinku nefronu związana z defektem wewnątrznerkowym bądź działaniem czynników pozanerkowych powoduje wrażliwość ciśnienia tętniczego na podaż sodu i staje się przyczyną nadciśnienia tętniczego. Uzyskano także argumenty, że nadciśnienie takie może być skutecznie leczone przez zablokowanie czynników powodujących zaburzenie mechanizmu natriurezy ciśnieniowej. Odkrycia te nasiliły poszukiwania genetycznych uwarunkowań cechy sodowrażliwości ciśnienia tętniczego, które mogłyby być podstawą rozwoju nadciśnienia, rozpoznawanego jako nadciśnienie pierwotne (samoistne). Wyniki najnowszych badań sugerują, że jest mało prawdopodobne, aby mutacje genów podjednostek b i g nabłonkowego kanału sodowego spełniały istotną rolę w patogenezie pierwotnego nadciśnienia tętniczego u ludzi. Wykazana początkowo dodatnia asocjacja między występowaniem allelu 344T w promotorowym odcinku genu syntazy aldosteronowej (CYP11B2) a pierwotnym nadciśnieniem tętniczym, sugerująca umiarkowany udział mutacji tego genu w rozwoju choroby (5), nie została potwierdzona w badaniach przeprowadzonych w Polsce (6).
Ostatnio Chiolero i wsp. (7) przedstawili dowody, że u osób z nadciśnieniem sodowrażliwym występuje zaburzenie czynności cewek proksymalnych nefronów polegające na nasilonej reabsorpcji sodu podczas żywienia dietą wysokosodową. Autorzy zaproponowali hipotezę, że osoby z nadciśnieniem sodowrażliwym wykazują niezdolność do adaptacji reabsorpcji sodu w cewkach proksymalnych nefronów podczas spożywania diety wysoko- lub niskosodowej. Hipoteza ta znajduje potwierdzenie w badaniach Barba i wsp. (8), którzy wykazali, że normotensyjni mężczyźni posiadający cechę sodowrażliwości ciśnienia tętniczego mają większą bezwzględną proksymalną reabsorpcję sodu podczas spożywania diety bogatosodowej w porównaniu z mężczyznami wykazującymi sodooporność ciśnienia tętniczego.
Jak wynika jednak z różnorodności mechanizmów, które mogą być przyczyną sodowrażliwości ciśnienia tętniczego, cecha ta może być wynikiem interakcji wielu uwarunkowań genetycznych z czynnikami środowiskowymi. W ostatnich latach zwrócono szczególną uwagę, że zmiany wewnątrznerkowych stężeń angiotensyny II, tlenku azotu (NO) i prostaglandyn (które mogą być uwarunkowane genetycznie), są odpowiedzialne za koordynację zmian hemodynamiki nerkowej i reabsorpcji sodu w cewkach nerkowych. Zaburzenie równowagi tych mechanizmów może powodować zmiany w natriurezie ciśnieniowej podobne do tych, jakie występują w rozwoju sodowrażliwego pierwotnego nadciśnienia tętniczego. Wykazano ostatnio, że całkowite wytwarzanie NO w warunkach podstawowych u chorych na nadciśnienie tętnicze jest zmniejszone w porównaniu z normotonikami, ale nie określono czy istnieje uwarunkowanie genetyczne tego zjawiska (9). Genetycznie uwarunkowana sodowrażliwość ciśnienia tętniczego może wiązać się z punktową mutacją w kodującym regionie G460W genu a-adducyny. Niektóre badania u zwierząt doświadczalnych jak i u ludzi sugerują, że obecność allelu W tego genu wiąże się z nasiloną reabsorpcją sodu, co prowadzi do rozwoju nad- ciśnienia tętniczego (10). Inne jednak badania przeprowadzone w Polsce nie potwierdzają tych spostrzeżeń (11). Cechę sodowrażliwości ciśnienia tętniczego wiąże się także z zaburzeniami działania przedsionkowego peptydu natriuretycznego (ANF), nerkowej kalikreiny, współczulnego układu nerwowego lub jego receptorów, insuliny i zaburzeniami błonowego transportu jonów, ale poszukuje się ciągle pewnych dowodów na rolę podłoża genetycznego w tych zaburzeniach.
W badaniach przeprowadzonych niedawno w Polsce uzyskano dowody, że u chorych z nadciśnieniem tętniczym mutacja T(r)C nukleotydu 2238 genu prekursora przedsionkowego peptydu natriuretycznego (ANP), prowadząca do eliminacji prawidłowego kodonu stop, wiąże się zarówno z cechą sodowrażliwości ciśnienia tętniczego jak i wyższymi stężeniami ANP w osoczu (12, 13). Wykazano także, iż polimorfizm w promotorowym odcinku genu ludzkiej kalikreiny nerkowej (HKLKI) wiąże się z nadciśnieniem tętniczym, cechą sodowrażliwości i zmniejszonym wydalaniem kalikreiny w moczu (14). W najnowszych, nieopublikowanych badaniach poświęconych poszukiwaniu związku między określonymi genotypami uwarunkowanymi polimorfizmami kilku genów-kandydatów (ANP, CYP11B2, HKLKI, I/D ACEI) wykazano możliwość określenia stopnia sodowrażliwości (to jest różnicy między wartościami średniego ciśnienia tętniczego na diecie bogato- i ubogosodowej) w postaci równania regresji u osób normotensyjnych (Ciechanowicz A., w przygotowaniu do druku). Wynik ten potwierdza sugestię, że połączenie występowania określonych alleli kilku genów silniej wiąże się z predyspozycją do występowania cechy sodowrażliwości ciśnienia tętniczego niż posiadanie mutacji alleli jednego z genów kandydatów.
Jako ważnego argumentu przemawiającego, że osoby sodowrażliwe mogą być genetycznie predysponowane do rozwoju nadciśnienia przyjmuje się wykazanie istotnie częstszego występowania cechy sodowrażliwości u młodych normotensyjnych potomków rodzin z dodatnim wywiadem nadciśnieniowym w porównaniu z osobami bez takiego obciążenia rodzinnego. Na podstawie przedstawionych danych można stwierdzić, że u pewnego odsetka osób cecha fenotypowa sodowrażliwości ciśnienia tętniczego może być związana z predyspozycją genetyczną uwarunkowaną polimorfizmem jednego lub częściej kilku genów kodujących białka uczestniczące w regulacji natriurezy ciśnieniowej. Chociaż w chwili obecnej trudno określić odsetek tych osób w populacji ogólnej, to można sądzić, że jest on raczej niewielki.
Badania doświadczalne dostarczyły dowodów na występowanie genetycznych uwarunkowań sodowrażliwości ciśnienia tętniczego. U niektórych szczepów szczurów na drodze krzyżowań wsobnych hodowli wyselekcjonowano grupy zwierząt różniące się wrażliwością na podawanie soli w pożywieniu. U uzyskanych w ten sposób szczurów sodowrażliwych żywienie dietą o dużej zawartości soli powodowało rozwój nadciśnienia tętniczego, w odróżnieniu od zwierząt sodoniewrażliwych. W obu grupach każda z tych cech była przekazywana w kolejnych pokoleniach wsobnej hodowli, stając się cechą uwarunkowaną genetycznie. Jeżeli jednak szczury podszczepu sodowrażliwego były od urodzenia żywione dietą o małej zawartości sodu, to nie powstało u nich nadciśnienie tętnicze, lub pojawiało się ono dopiero pod koniec życia. Udowodniono w ten sposób, że fenotypowa cecha sodowrażliwości bądź sodoniewrażliwości ciśnienia tętniczego może być uwarunkowana genetycznie, a o wystąpieniu nadciśnienia tętniczego u osobników sodowrażliwych decyduje czynnik środowiskowy jakim jest duża podaż sodu w diecie.
Liczne badania przeprowadzone na pierwszym eksperymentalnym modelu sodowrażliwości ciśnienia tętniczego jakim są szczury Dahla umożliwiły identyfikację dość dużej ilości miejsc (lokus) zlokalizowanych na chromosomach 1, 2, 3, 5, 7, 9, 10, 13 i 17, zawierających najprawdopodobniej geny odpowiedzialne za sodowrażliwość (15). Nie udało się jednak określić, które z tych miejsc są ściśle związane z sodoniewrażliwością, ponieważ u szczurów Dahla, poza cechami sodowrażliwości bądź sodowrażliwości rozwija się z wiekiem samoistne nadciśnienie tętnicze, nawet przy ograniczonej podaży soli. W innych modelach eksperymentalnych potencjalne geny, związane z sodowrażliwością były w znacznej części zlokalizowane odmiennie, co sugeruje dużą heterogenność cechy.
Nowym modelem doświadczalnego nadciśnienia sodowrażliwego i sodoopornego stały się szczury Sabra (16). W tym modelu u zwierząt sodowrażliwych (SBH/y) genetycznie uwarunkowane nadciśnienie tętnicze ujawnia się wyłącznie w warunkach obciążenia sodem, zaś przy ograniczeniu podaży sodu nie rozwija się u nich nadciśnienie samoistne. W modelu tym zidentyfikowano 3 prawdopodobne geny warunkujące sodowrażliwość. Te genetyczne uwarunkowania dotyczyły fizjologicznych mechanizmów związanych z regulacją gospodarki sodowej, takich jak układ tlenku azotu (NO), argininowej wazopresyny i nabłonkowego kanału sodowego. Określono także 3 przypuszczalne genetyczne miejsca zlokalizowane na chromosomach 1 (dwa miejsca) i 17, które istotnie wpływają na sodowrażliwość i/lub sodooporność, ale nie zidentyfikowano poszczególnych genów w obrębie tych miejsc. Ponadto wykryto niezidentyfikowaną do końca specyfikę związaną z płcią, która wpływa na rolę jaką spełniają geny sodowrażliwości w rozwoju nadciśnienia tętniczego a także skomplikowany schemat dziedziczenia tej cechy. Ekstrapolacja wyników uzyskanych w tym szczególnym modelu sodowrażliwości nadciśnienia tętniczego na możliwe mechanizmy działające u ludzi musi być niezwykle ostrożna.
Podstawą do nowego kierunku rozważań nad przyczynami sodowrażliwości ciśnienia tętniczego stały się opublikowane ostatnio wyniki badań Keller i wsp. (17). Autorzy ci przedstawili bezpośrednie dowody, że liczba nefronów u chorych na pierwotne nadciśnienie tętnicze rasy białej jest istotnie mniejsza niż w starannie dobranej grupie osób normotensyjnych. W badaniach wykorzystano trójwymiarową stereologiczną metodę do porównania liczby i objętości kłębuszków nerkowych u 10 osób w wieku 35-50 lat z dodatnim wywiadem w kierunku nadciśnienia tętniczego lub/i z przerostem lewej komory serca z 10 osobami normotensyjnymi dobranymi pod względem płci, wieku, wysokości i masy ciała. Materiał do niezwykle dokładnie przeprowadzonych badań uzyskano od osób, które zginęły w wypadkach. Wykazano, że osoby z nadciśnieniem tętniczym miały istotnie mniejszą liczbę kłębuszków nerkowych przypadających na nerkę (mediana 702 379) w porównaniu z osobami normotensyjnymi (mediana 1429 200). W przedstawionych wynikach zwraca uwagę, że za wyjątkiem jednej osoby, wszystkie pozostałe z nadciśnieniem tętniczym miały mniejszą liczbę kłębuszków nerkowych niż odpowiadające im osoby normotensyjne. Ponadto osoby z nadciśnieniem tętniczym wykazywały istotnie większą objętość kłębuszków nerkowych niż osoby normotensyjne (p <0,001), a w porównywanych grupach nie występowało nakładanie się indywidualnych wartości. W nerkach osób z nadciśnieniem tętniczym znaleziono tylko pojedyncze obumarłe kłębuszki nerkowe i przeprowadzono dowód, że za zmniejszoną liczbę kłębuszków nerkowych w tych przypadkach nie jest odpowiedzialny zanik kłębuszków wywołany chorobą. Wyniki tego badania potwierdzają opublikowaną wcześniej tezę Brennera (18, 19), że liczba nefronów, która jest zdeterminowana w okresie życia płodowego osoby, staje się ważną determinantą rozwoju nie tylko nadciśnienia, ale także zaburzeń sercowo-naczyniowych w życiu dorosłym. Keller i wsp. (17) zwracają uwagę, że nie zostało wyjaśnione dotychczas, czy zmniejszona liczba nefronów u osób z nadciśnieniem tętniczym jest spowodowana przez czynniki genetyczne, czy też przez czynniki środowiskowe. Otwiera to drogę do dalszych badań, nawet przy uwzględnieniu istniejących dowodów na istniejącą odwrotną zależność między masą urodzeniową osoby i wysokością ciśnienia tętniczego w życiu dorosłym. W badaniach doświadczalnych uzyskano dowody, że niedożywienie białkowe w okresie ciąży powoduje występowanie zmniejszonej liczby nefronów w nerkach potomstwa nawet o 20% w stosunku do przeciętnej liczby nefronów u potomstwa prawidłowo odżywianych matek. Wydaje się jednak mało prawdopodobne aby wyłącznie niedożywienie spowodowało wykazaną przez Keller i wsp. (17) różnicę w liczbie nefronów między osobami z samoistnym nadciśnieniem tętniczym i dobranymi osobami normotensyjnymi. Bardziej prawdopodobne wydaje się założenie, że wrodzona liczba nefronów zależy zarówno od wpływu czynników genetycznych jak i środowiskowych oraz ich wzajemnych interakcji w rozwoju płodowym. Na możliwość wpływu czynników genetycznych warunkujących wrodzoną liczbę nefronów zwraca uwagę Ingelfinger i wsp. (20), ale brak dotychczas bezpośrednich danych u ludzi potwierdzających tę tezę.
Mechanizm, za pośrednictwem którego wrodzona zmniejszona liczba nefronów wpływa na rozwój nadciśnienia tętniczego nie został jeszcze wyjaśniony. Najprostsze wydaje się założenie, że wszystkie mechanizmy uczestniczące w rozwoju nadciśnienia tętniczego powodują jego wcześniejsze ujawnienie się u osoby z wrodzoną zmniejszoną liczbą nefronów. Zwraca jednak uwagę, że wyniki pracy Keller i wsp. (17) udokumentowały, iż mniej liczebne kłębuszki u osób z nadciśnieniem tętniczym różnią się morfologicznie od kłębuszków osób normotensyjnych. Kłębuszki osób z nadciś- nieniem tętniczym charakteryzowały się zwiększoną o 133% objętością w porównaniu z osobami normotensyjnymi co dowodzi, że uległy one adaptacyjnej hipertrofii, podobnie jak to następuje przy zmniejszonej liczbie nefronów z innych przyczyn, np. przy ich niszczeniu przez przewlekłą kłębuszkową nefropatię. W takich warunkach reabsorbcja sodu w również przerosłej cewce bliższej ulega nasileniu, natomiast dostosowanie wydalania sodu do jego podaży zachodzi głównie w pętli Henlego i w dystalnych odcinkach nefronów, a ciśnienie tętnicze staje się sodowrażliwe. W chwili obecnej brak bezpośrednich dowodów u ludzi, że wrodzona zmniejszona liczba nefronów wiąże się z cechą sodowrażliwości ciśnienia tętniczego.
Dowody takie istnieją natomiast u zwierząt doświadczalnych z genetycznie uwarunkowaną zmniejszoną liczbą nefronów. Szczury szczepu Munich Wistar Frömter (MWF) cechują się zmniejszoną o 30-50% liczbą nefronów w porównaniu ze szczurami kontrolnymi. U zwierząt tych rozwija się samoistne nadciśnienie tętnicze i zwiększa wydalanie albumin w moczu. Żywienie szczurów MWF dietą wysokosodową powoduje wybitne nasilenie nadciśnienia tętniczego jak również znaczne zwiększenie albuminurii w porównaniu z żywieniem dietą niskosodową (21). U zwierząt kontrolnych z prawidłową liczbą nefronów nie rozwijało się nadciśnienie tętnicze oraz nasilona albuminuria, niezależnie od małej czy dużej zawartości sodu w karmie. Na tej podstawie uzyskano dowody, że wrodzona zmniejszona liczba nefronów może prowadzić do rozwoju sodowrażliwego nadciśnienia tętniczego i upośledzonej czynności nerek (21). Bezpośrednie udokumentowanie, że tak jest również u ludzi, powinno wnieść nowe elementy do wyjaśnienia podłoża zjawiska sodowrażliwości ciśnienia tętniczego.
We wcześniej opublikowanych pracach (22, 23) przedstawiona została hipoteza S. Czekalskiego i A. Ciechanowicza, że występowanie i nasilenie zjawiska sodowrażliwości ciśnienia tętniczego odzwierciedla liczbę czynnych nefronów. Schemat ilustrujący ideę tej hipotezy przedstawiono na rycinie 1.
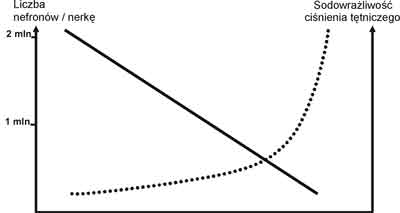
Ryc. 1. Występowanie i nasilenie zjawiska sodowrażliwości ciśnienia tętniczego odzwierciedla liczbę czynnych nefronów.
Hipoteza ta wymaga udowodnienia, ale liczne argumenty przytoczone we wcześniejszych publikacjach przemawiają na jej korzyść, nie wykluczając wpływu innych, w tym genetycznych czynników determinujących cechę sodowrażliwości ciśnienia tętniczego u ludzi (24). W oparciu o powyższe dane można stwierdzić, że cecha sodowrażliwości ciśnienia tętniczego u znacznego odsetka osób normotensyjnych jest uwarunkowana genetycznie, pod wpływem działania bliżej nieokreślonych czynników działających w okresie życia płodowego i wpływających na wrodzoną zmniejszoną liczbę nefronów i/lub mechanizmy natriurezy ciśnieniowej.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Luft F.C. et al.: Cardiovascular and humoral responses to extremes of sodium intake in normal black and white men. Circulation 1979, 60, 697-706.
2. Szczepańska E.: Rola nerek w regulacji ciśnienia tętniczego, w: Nadciśnienie tętnicze, red: A. Januszewicz, W. Januszewicz, E. Szczepańska-Sadowska, M. Sznajderman, Wydawnictwo Medycyna Praktyczna, Kraków 2000, 271-280
3. Czekalski S.: Sodowrażliwość a nadciśnienie tętnicze. W: Nad- ciśnienie tętnicze, red: A. Januszewicz, E. Szczepańska-Sadowska, M. Sznajderman, Wydawnictwo Medycyna Praktyczna, Kraków 2000, 397-401.
4. Zaremba J.: Genetycznie uwarunkowane nadciśnienie tętnicze u ludzi, w: Nadciśnienie tętnicze, red: A. Januszewicz, W. Januszewicz, E. Szczepańska-Sadowska, M. Sznajderman, Wydawnictwo Medycyna Praktyczna, Kraków 2000, 55-59.
5. Brand E. et al.: Structural analysis and evaluation of the aldosterone synthase gene in hypertension. Hypertension, 1998, 32, 198-204.
6. Wrona A. i wsp.: Warianty promotora genu syntazy aldosteronu (CYP11B2) i sodowrażliwość ciśnienia tętniczego. Pol. Arch. Med. Wew. 2004, CXI, 191-198.
7. Chiolero A. et al.: Renal determinants of the salt sensitivity of blood pressure. Nephrol Dial Transplant 2001, 16, 452-458.
8. Barba G. et al.: Renal function and blood pressure response to dictary salt restriction in normotensive men. Hypertension 1996, 27, 1160-1164.
9. Forte P. et al.: Basal nitric oxide synthesis in essential hypertension. Lancet 1997, 349, 837-842. Kamitani A., Wong Z.Y.H., Fraser R. i wsp.: Human a-adducin gene, blood pressure, and sodium metabolism. Hypertension 1998, 32, 138-143.
10. Ciechanowicz A. et al.: Lack of association between Gly450Trp polymorphism of alpha- adducing gene and salt sensitivity of blood pressure in polish hypertensives. Kidney Blood Press 2001, 24, 201-206.
11. Widecka K. i wsp.: Analiza polimorfizmów SmaI (Hpa II) I Sca I genu prekursora przedsionkowego peptydu natriuretycznego (ANP) u chorych z samoistnym nadciśnieniem tętniczym. Pol Arch Med Wewn 1998, 7, 27-34.
12. Ciechanowicz A. i wsp.: Mutacja (T(r)C) nukleotydu 2238 genu prekursora przedsionkowego peptydu natriuretycznego (ANP) i niejednorodność sodowrażliwego nadciśnienia tętniczego. Pol Arch Med. Wewn. 1997, 12, 501-503.
13. Placha G. et al.: DNA polymorphism in promoter of human renal kallikrein gene (HKLKI) is associated with hypertension, salt sensitivity and urinary kallikrein excretion. J Am Soc Nephrol 2001, 12, A2430.
14. Rapp J.P.: The genetics of hypertension in Dahl rats. w: Experimental and genetic models of hyperetnsion. Red.: D. Ganten, W. De Jong, Elsevier Science B.V., Amsterdam 1994.
16. Yagil Y., Yagil C.: Genetic basis of salt-susceptibility in the Sabva rat model of hypertension. Kidney Intern. 1998, 53, 1493-1500.
17. Keller G. et al.: Nephron number in patients with primary hypertension. New Engl J Med. 2003, 348, 101-108.
18. Brenner BM. et al.: Glomeruli and blood pressure. Less of one, more the other? Am J Hypertens 1988, 1, 335-347.
19. Brenner BM, Chertow GM.: Congential oligonephropathy and the etiology of adult hypertension and progressive renal injury. Am J Kidney Dis. 1994, 23, 171-175.
20. Ingelfinger J.R.: Is microanatomy destiny? New Engl J Med. 2003, 348, 99-100.
21. Kreuz R. et al.: Effect of high NaCl diet on sponaneous hypertension in a genetic model with reduced nephron number. J Hypertens 2000, 18, 777-782.
22. Czekalski S, Oko A.: Czy nadciśnienie tętnicze pierwotne jest w większości przypadków związane z wrodzoną zmniejszoną liczbą nefronów? Nadciśnienie Tętnicze 2003, 7, 1-6.
23. Czekalski S., Pawlaczyk K.: Nowe poglądy na patogenezę pierwotnego nadciśnienia tętniczego i nadciśnienia w chorobach nerek. Nefrologia i Dializoterapia Polska, 2003 (w druku).
24. Ciechanowicz A.: Molekularne podłoże nadciśnienia tętniczego - przegląd genów kandydatów, W: Genetyka chorób układu krążenia, red: A. Ciechanowicz, A. Januszewicz, W. Januszewicz, W. Rużyłło. Medycyna Praktyczna, Kraków 2002, 191-198.
25. Johnson R.J., Herreva-Acosta J., Schreiner G.F., Rodriquez-Iturbe. B: Subtle acquired renal injury as a mechanism of salt-sensitive hypertension. New Engl J Med 2002, 346, 913-923.
26. Ardiles L.G. et al.: Renal kalikrein-kinin system damage and salt sensitivity: Insights from experimental models. Kidney Int 2003, 64, suppl 86, S2-S8.
27. Rocchini A.P. et al.: The effect of weight loss on the sensitivity of blood pressure to sodium in obese adolescents. N Engl J Med. 1989, 321 580-585.
28. Young J.B., Landsberg L.: Stimulation of the sympathetic nervous system during sucrose feeding. Nature 1977, 269, 615-618.
29. Schwartz J.H. et al.: Effects of dietary fat on sympatheic nervous system activity in the rat. J Clin Invest 1983, 72, 361-369.
30. Landsberg L.: Obesity - related hypertension as a metabolic disorder, in: Hypertension, eds: W. Oparil, M.A. Weber, W.B. Saunders comp., Philadelphia 2000, 118-124.
31. Angielski S.: Insulina, w: Nadciśnienie Tętnicze, red.; A. Januszewicz, W. Januszewicz, E. Szczepańska-Sadowska, M. Sznajderman, Wydawnictwo Medycyna Praktyczna, Kraków 2000, 203-206.
32. Czekalski S, Ciechanowicz A: Nadciśnienie tętnicze nerkopochodne - znane i nieznane. Nefrologia i Nadciśnienie Tętnicze 2003, s/4, 7-10.
33. Morimoto A. et al.: Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet 1997, 350, 1734-1737.
34. Ardiles L.G. et al.: Effect of mycophenolate mofetil on kallikrein expression in the kidney of 5/6 nephrectomized rats. Kidney Blood Press Res 2002, 25, 289-295.
35. Wolf W.C. et al.: Human tissue kallikrein gene delivery attemates hypertension, renal injury, and cardiac remodeling in chronic renal failure. Kidney Int 2000, 58, 730-739.
