Hiperfosfatemia – nowy czynnik w patogenezie powikłań sercowo-naczyniowych u chorych z niewydolnością nerek
Hyperphosphataemia – a new factor in the pathogenesis of cardio-vascular complications in uraemic patients
z Katedry i Kliniki Nefrologii, Endokrynologii i Chorób Przemiany Materii, Śląskiej Akademii Medycznej w Katowicach
Kierownik Kliniki: prof. dr hab. Andrzej Więcek
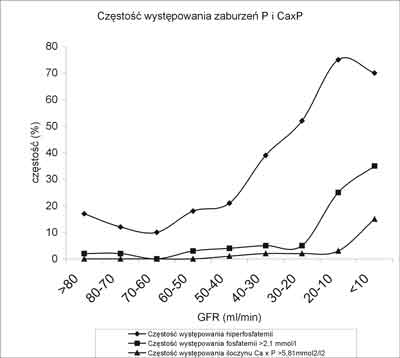
Nerki odgrywają kluczową rolę w utrzymaniu zrównoważonego bilansu fosforanowego ustroju. Już we wczesnym stadium niewydolności tego narządu, tj. przy obniżeniu filtracji kłębuszkowej (GFR) poniżej 50ml/min, wyczerpują się zdolności kompensacyjne dla wydalania fosforanów. Następstwem tego jest wzrost stężenia fosforanu w surowicy (1). W okresie schyłkowej niewydolności nerek częstość występowania hiperfosfatemii zarówno wśród chorych nieleczonych nerkozastępczo (również populacji polskiej) (2) (ryc. 1), jak i u leczonych powtarzanymi dializami (3) sięga blisko 70%.
Ryc. 1. Częstość występowania zaburzeń w gospodarce wapniowo-fosforanowej w populacji polskiej wśród chorych nieleczonych nerkozastępczo (2).
Jeszcze do połowy lat 90. ubiegłego stulecia uważano, że hiperfosfatemia prowadzi do wtórnej nadczynności przytarczyc, osteodystrofii nerkowej i zwapnień w tkankach okołostawowych jedynie pośrednio poprzez hamowanie aktywności 1a-hydroksydazy i syntezy kacytriolu w nerkach (ograniczenie wchłaniania wapnia z przewodu pokarmowego) oraz poprzez zmniejszenie frakcji wapnia zjonizowanego w osoczu – hipokalcemię. Jednak w ostatnich latach w badaniach in vitro oraz w badaniach doświadczalnych u zwierząt udowodniono, że hiperfosfatemia wpływa na przytarczyce również bezpośrednio, niezależnie od stężenia kalcytriolu oraz wapnia w osoczu, zwiększając zarówno wydzielanie PTH jak i pobudzając ich rozrost (4). Hiperfosfatemia poprzez interakcję z białkiem cytozolowym AUF-1 stabilizuje mRNA dla PTH (mechanizm posttrankrypcyjny) i zwiększa przez to syntezę tego hormonu (5). Rozrost przytarczyc, wydaje się być następstwem nasilonej pod wpływem hiperfosfatemii ekspresji TGFa (transformujący czynnik wzrostu alfa) (6) i hamowania syntezy białka p21 – inhibitora kinaz cytokinozależnych (7). Ograniczenie podaży fosforanów w diecie prowadzi natomiast do obniżenia syntezy i stężenia PTH w surowicy (8, 9).
W badaniach epidemiologicznych przeprowadzonych przez Block´a i wsp. wykazano po raz pierwszy, że u chorych hemodializowanych ze stężeniem fosforu w osoczu przekraczającym 6,5 mg% (2,1 mmol/l) ryzyko zgonu jest o 27% wyższe w porównaniu do chorych z normofosfatemią (3). Autorzy ci wykazali również, że wzrost iloczynu Ca x P powyżej 72 mg2/dl2 (5,81 mmol2/l2) zwiększa roczną śmiertelność chorych o 34% w porównaniu z grupą z wartościami tego iloczynu od 43 do 52 mg2/dl2 (4,47-4,20 mmol2/l2). Należy podkreślić, że ryzyko zgonu związane z hiperfosfatemią było niezależne od stężenia parathormonu w surowicy. Jednocześnie autorzy nie stwierdzili zależności pomiędzy stężeniem wapnia całkowitego w osoczu a globalnym ryzykiem zgonu w badanej grupie. Bardziej szczegółowa analiza przyczyn zgonów tych chorych wskazała, że ryzyko zgonu z powodu choroby niedokrwiennej serca było wyższe o 41% w porównaniu do wszystkich przyczyn zgonu (wzrost o 21%) (10). Jak już na początku wspomniano nadmierna retencja fosforanów jest częstym następstwem schyłkowej niewydolności nerek. Block i wsp. stwierdzili, że częstość hiperfosfatemii wśród chorych hemodializowanych pomimo stosowania leków wiążących fosforany w przewodzie pokarmowym sięga 60%, a u 39% wartości przekraczają 6,5 mg/dl (>2,1 mmol/l) (11). Również badania przeprowadzone wśród chorych hemodializowanych w krajach Europy (Włochy i Hiszpania) potwierdzają wysoką, wynoszącą około 25% częstość występowania znacznej hiperfosfatemii (>6,5 mg/dl). Natomiast podwyższone wartości iloczynu CaxP (>60 mg2/dl2) stwierdzono u blisko jednej trzeciej badanych (12, 13). O wartości CaxP decyduje głównie stężenie fosforu (r2= 0,914), natomiast kalcemia ma znaczenie drugorzędne (r2= 0,229) (14).
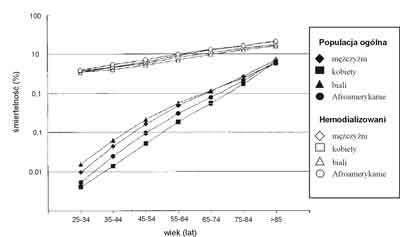
Pomimo poprawiającej się opieki nefrologicznej nad chorymi niewymagającymi jeszcze leczenia nerkozastępczego oraz doskonalenia metod samej dializoterapii, śmiertelność chorych dializowanych pozostaje ciągle na tym samym poziomie co w latach poprzednich (15). Szacuje się, że średni oczekiwany czas przeżycia chorych rozpoczynających przewlekłą hemodializoterapię w wieku 40-44 lat wynosi jedynie 7-10 lat (15). Główną przyczyną zgonów chorych leczonych powtarzanymi hemodializami są choroby układu krążenia (15). Śmiertelność w grupie chorych dializowanych w wieku od 45-54 lat jest ponad sto razy większa niż w populacji ogólnej w podobnym przedziale wiekowym (15). Co więcej, ryzyko zgonu z powodu powikłań sercowo-naczyniowych w tej grupie jest nawet wyższe niż dla ludzi w wieku podeszłym, u których nie stwierdzono niewydolności nerek (16) (ryc. 2). Tak zwiększone ryzyko zgonu z powodu chorób układu krążenia wiąże się zarówno z występowaniem u tych chorych tzw. klasycznych czynników ryzyka, takich jak: nadciśnienie tętnicze, cukrzyca, otyłość trzewna, dyslipidemia, palenie tytoniu, czy hiperhomocysteinemia. Dominujące znaczenie mają jednak czynniki ryzyka specyficzne dla chorych przewlekle dializowanych. Na szczególną uwagę zasługuje niedokrwistość, przeciążenie układu krążenia z powodu hiperwolemii i nagłych zmian wolemii w trakcie hemodializy, obecności zespoleń tętniczo-żylnych (dostęp do hemodializy); stres oksydacyjny, hiperkatabolizm, kumulacja toksyn mocznicowych, przewlekłe stany zapalne oraz zaburzenia gospodarki wapniowo-fosforowej (3, 10, 16, 17, 18). Warto zauważyć, że wyniki najnowszych badań sugerują występowanie związku pomiędzy iloczynem wapniowo-fosforanowym a nasileniem stanu zapalnego – stężeniem białka C-reaktywnego (CRP) w surowicy. Obniżenie wartości iloczynu Ca x P poniżej 55 mg2/dl2 powoduje natomiast znaczne obniżenie stężenia CRP w surowicy u chorych hemodializowanych (19, 20).
Ryc. 2. Śmiertelność z powodu chorób układu krążenia w populacji ogólnej oraz wśród chorych dializowanych (16).
Modyfikowalny charakter hiperfosfatemii oraz podwyższonego iloczynu Ca x P jako czynników przyczyniających się do zwiększenia śmiertelności, w szczególności z powodu chorób układu krążenia, ma duże znaczenie praktyczne. Zwapnienie tkanek miękkich, w tym ścian tętnic (21, 22, 23), mięśnia sercowego (24) czy zastawek serca (21, 22, 25) są nie tylko znacznie bardziej nasilone u chorych na mocznicę niż w populacji ogólnej, lecz obserwuje się je nieporównywalnie wcześniej. Już u chorych rozpoczynających leczenie nerkozastępcze stopień zaawansowania zmian naczyniowych jest istotnie większy niż w populacji ogólnej. Co więcej, badania autopsyjne potwierdzają, że zwapnienia w naczyniach u chorych na schyłkową niewydolność nerek lokalizują się głównie w błonie środkowej, a nie w błonie wewnętrznej jak u chorych ze zmianami miażdżycowymi w populacji ogólnej bez niewydolności nerek (26). Jest to szczególnie widoczne u pacjentów długotrwale dializowanych, którzy rozpoczynali leczenie nerkozastępcze w młodszym wieku (27), Braun i wsp. używając tomografii wiązką elektronów (EBT) wykazali, że częstość występowania kalcyfikacji naczyń wieńcowych u osób w wieku 40-49 lat jest aż pięciokrotnie wyższa w grupie hemodializowanych niż wśród chorych na chorobę niedokrwienną serca z prawidłową czynnością wydalniczą nerek (21). Goodman i wsp., również stosując metodę EBT, obserwowali zwapnienia w tętnicach wieńcowych aż u 87% hemodializowanych dwudziestolatków i jedynie u 5% ich zdrowych rówieśników (22). Autorzy ci nie stwierdzili występowania kalcyfikacji naczyń wieńcowych jedynie w grupie dializowanych chorych w wieku poniżej 20 roku życia (22). Nasilenie zwapnień w obrębie ściany naczyń było większe u osób starszych, dłużej leczonych nerkozastępczo, z wyższymi stężeniami fosforu nieorganicznego w osoczu i wartościami iloczynu CaxP oraz przyjmujących wyższe dawki octanu wapnia (22). Natomiast stężenie parathormonu oraz cholesterolu całkowitego było u chorych hemodializowanych ze zmianami naczyniowymi niższe niż u tych, u których nie stwierdzano zwapnień w naczyniach. Prawie u wszystkich chorych ze zwapnieniami naczyń wieńcowych obserwowano dalszą progresję zmian (22). Wpływ zaburzeń gospodarki wapniowo-fosforanowej na stopień nasilenia zwapnień naczyń wieńcowych oraz częstość występowania choroby niedokrwiennej serca badali ostatnio również Raggi i wsp. (23). Badania te jednoznacznie potwierdzają ścisłą zależność pomiędzy stopniem zwapnienia ścian naczyń (mierzonym przy użyciu EBT) a częstością występowania chorób układu krążenia. W grupie o najwyższych wartościach wskaźnika uwapnienia naczyń (Agatson score>1000) incydenty wieńcowe i/lub zmiany w obrębie naczyń obwodowych występowały u ponad 80% badanych chorych, natomiast przy wartości wskaźnika Agatson mniejsze niż 400 objawy takie występowały jedynie u około 20% chorych. Przeprowadzona analiza wieloczynnikowa wykazała, że wzrost stężenia wapnia całkowitego, oraz fosforu w osoczu o każde 1 mg/dl ma taki sam wpływ na stopień kalcyfikacji ścian naczyń wieńcowych jak odpowiednio 5 i 2,5 roku leczenia powtarzanymi hemodializami. Zwapnienia zastawek mitralnych i aortalnych obserwowano odpowiednio u 45% i 34% chorych, a co piąty badany miał obie zastawki zwapniałe (23). Największy wpływ na występowanie zwapnień zastawek mitralnych wśród hemodializowanych chorych ma podwyższony iloczyn CaxP oraz wiek pacjenta (25). Co ciekawe, zależności te nie znalazły potwierdzenia w odniesieniu do zastawek aortalnych (25). Dowodów na ścisłą zależność pomiędzy wartością iloczynu CaxP a obecnością zwapnień w naczyniach dostarczyli również Shigegatsu i wsp., którzy wykazali, że wartości iloczynu CaxP> 65 mg2/dl2 wiążą się z obecnością zwapnień w łuku aorty (14). Wykazano również, że chorzy, u których obserwuje się zaawansowaną kalcyfikację naczyń (zarówno aorty jak i naczyń wieńcowych) charakteryzują się znacząco mniejszą gęstością mineralną kości. Ten fakt sugeruje istnienie związku pomiędzy chorobami układu krążenia a ostedystrofią nerkową (28), a w szczególności postacią adynamiczną tej choroby (29). Jednak z klinicznego punktu widzenia najistotniejszy jest fakt, iż wzrost uwapnienia naczyń wiąże się ze zwiększonym ryzykiem zgonu (30).
1. Hsu C.Y., Chertow GM.: Elevations of serum phosphorus and potassium in mild to moderate chronic renal insufficiency. Nephrol Dial Transplant 2002, 17: 1419-1425
2. Zarzecki M. i wsp.: Stopień niewydolności nerek a częstość występowania niedokrwistości, zaburzeń gospodarki wapniowo-fosforanowej oraz kwasicy metabolicznej. Pol Arch Med Wewn. (w druku).
3. Block GA. et al.: Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: a national study. Am J Kidney Dis 1998, 31: 607-617.
4. Slatopolsky E. et al.: Phosphorus restriction prevents parathyroid gland growth. High phosphorus directly stimulates PTH secretion in vitro. J Clin Invest. 1996, 97: 2534-2540.
5. Kilav R, Silver J, Naveh-Many T. A conserved cis-acting element in the parathyroid hormone 3´-untranslated region is sufficient for regulation of RNA stability by calcium and phosphate. J Biol Chem. 2001, 276: 8727-8733.
6. Dusso A.S. et al.: p21(WAF1) and transforming growth factor-alpha mediate dietary phosphate regulation of parathyroid cell growth. Kidney Int. 2001, 59: 855-865.
7. Sherr C.J., Roberts J.M.: CDK inhibitors: positive and negative regulators of G1- phase progression. Genes Dev. 1999; 13: 1501-1512.
8. Kilav R. et al.: Parathyroid hormone gene expression in hypophosphatemic rats. J Clin Invest. 1995; 96: 327-333.
9. Combe C,, Aparicio M.: Phosphorus and protein restriction and parathyroid function in chronic renal failure. Kidney Int. 1994, 46: 1381-1386.
10. Ganesh S.K. et al.: Association of elevated serum PO4, Ca X PO4 product, and parathyroid hormone with cardiac mortality risk in chronic hemodialysis patients. J Am Soc Nephrol 2001, 12: 2131-2138.
11. Block G.A.: Prevalence and clinical consequences of elevated Ca x P product in hemodialysis patients. Clin Nephrol. 2000, 54: 318-324.
12. Gallieni M. et al.: Calcium, phosphate, and PTH levels in the hemodialysis population: A multicenter study. J Nephrol 2002, 15: 165-170.
13. Diaz Corte C. et al.: y los centros colaboradores del Estudio Multicentrico sobre Osteodistrofia Renal. Marcadores metabnlicos nseos y uso de vitamina D en dirlisis. Encuesta multicentrica. Nefrologia 2000, 20: 244-253.
14. Shigematsu T. et al.: Phosphate overload accelerates vascular calcium deposition in end-stage renal disease. Nephrol Dial Transplant 2003, 18 (suppl 3): iii86-iii89.
15. USRDS 1998. Annual data report. US Department of Health and Human Services. The National Institutes of Health. National Institiute of Diabetes and Digestive and Kidney Diseases. Bethesda, 1998.
16. Foley R.N. et al.: The impact of anemia on cardiomyopathy, morbidity and mortality in end-stage renal disease. Am J Kidney Dis 1996, 28: 53-61.
17. Avorn J. et al.: Delayed nephrologist referral and inadequate vascular access in patients with advanced chronic kidney failure. J Clin Epidemiol. 2002, 55: 711-716.
18. Samouilidou E.C. et al.: Oxidative stress markers and C-reactive protein in end-stage renal failure patients on dialysis. Int Urol Nephrol. 2003, 35: 393-397.
19. Movilli E. et al.: Correcting high calcium - phosohate product improves inflammatory status in hemodialysis patients: a prospective study. SP 255, ERA-EDTA XLI Congress, May 15-18 2004 Lisbon, Portugal.
20. Movilli E. et al.: High calcium - phosohate product worsnes inflammatory status in hemodialysis patients. SP 258, ERA-EDTA XLI Congress, May 15-18 2004 Lisbon, Portugal.
21. Braun J. et al.: Electron beam computed tomography in the evaluation of cardiac calcification in chronic dialysis patients. Am J idney Dis 1996, 27: 394-401.
22. Goodman W.G. et al.: Coronary-Artery Calcificaion in Youn Adults with End-Stage-Renal-Disease Who are Undergoing Dialsis. N Engl J Med 2000, 20: 1478-1483.
23. Raggi P. et al.: Cardiac Calcyfication in Adult Hemodialysis Patients. A link Between End-Stage-Renal-Disease and Cardiovascular Disease. J Am Col Cardiol 2002, 39: 695-701.
24. Rostand S.G. et al.: Myocardial calcification and cardiac dysfunction in chronic renal failure. AM J Med 1988, 85: 651-657
25. Riberio S. et al.: Cardiac valve calcification in haemodialysis patients: role of calcium-phospate metabolism. Nephrol Dial transplant 1998, 13: 2037-2040.
26. Schwarz U. et al.: Morphology of coronary atherosclerotic lesions in patients with end-sage renal failure. Nephrol Dial Transplant 2000, 15: 218-223.
27. London G.M. et al.: Arterial media calcification in end-stagr renal disease: Impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant 2003, 18: 1731-1740.
28. Raggi P. et al.: The relationship of trabecular bone density to coronary and aortic calcificaction. MP 256. ERA-EDTA XLI Congress, May 15-18 2004 Lisbon, Portugal.
29. London G.M. et al.: Arterial calcifications and bone histomorphometry in end-stage renal disease. J Am Soc Nephrol. 2004, 15: 1943-1951.
30. Blacher J. et al.: Arterial calcification, arterial stiffness, and cardiovascular risk in end-stage-renal-disease. Hypertension 2001, 38: 938-942.
31. Chen N.X., Moe S.M.: Vascular calcification in chronic kidney disease. Semin Nephrol. 2004, 24: 61-68.
32. Ahmed S. et al.: Calciphylaxis is associated with hyperphosphatemia and increased osteopontin expression by vascular smooth muscle cells. Am J Kidney Dis. 2001, 37: 1267-1276.
33. Shanahan C.M. et al.: Medial localization of mineralization-regulating proteins in association with Monckeberg´s sclerosis: evidence for smooth muscle cell-mediated vascular calcification. Circulation. 1999, 100: 2168-2176.
34. Moe S.M. et al.: Medial artery calcification in ESRD patients is associated with deposition of bone matrix proteins. Kidney Int. 2002, 61: 638-647.
35. Zebboudj A.F. et al.: Matrix GLA protein and BMP-2 regulate osteoinduction in calcifying vascular cells. J Cell Biochem. 2003, 90: 756-765.
36. Jono S. et al.: Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000, 87: E10-17.
37. Yoshida T. et al.: Mediation of unusually high concentrations of 1,25-dihydroxyvitamin D in homozygous klotho mutant mice by increased expression of renal 1alpha-hydroxylase gene. Endocrinology. 2002, 143: 683-689.
38. Kuro-o M. et al.: Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997, 390: 45-51.
39. Kawaguchi H. et al.: Independent impairment of osteoblast and osteoclast differentiation in klotho mouse exhibiting low-turnover osteopenia. J Clin Invest. 1999, 104: 229-237.
40. Suga T. et al.: Disruption of the klotho gene causes pulmonary emphysema in mice. Defect in maintenance of pulmonary integrity during postnatal life. Am J Respir Cell Mol Biol. 2000, 22: 26-33.
41. Okada S. et al.: Impairment of B lymphopoiesis in precocious aging (klotho) mice. Int Immunol. 2000, 12: 861-871.
42. Takeshita K. et al.: Sinoatrial node dysfunction and early unexpected death of mice with a defect of klotho gene expression. Circulation. 2004, 109: 1776-1782.
43. Kido S. et al.: Identification of regulatory sequences and binding proteins in the type II sodium/phosphate cotransporter NPT2 gene responsive to dietary phosphate. J Biol Chem. 1999, 274: 28256-28263.
44. Morishita K. et al.: The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. J Nutr. 2001, 131: 3182-3188.
45. Koh N. et al.: Severely reduced production of klotho in human chronic renal failure kidney. Biochem Biophys Res Commun. 2001, 280: 1015-1020.
46. Luo G. et al.: Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997, 386: 78-81.
47. Jahnen-Dechent W. et al.: Cloning and targeted deletion of the mouse fetuin gene. J Biol Chem. 1997; 272: 31496-31503.
48. Lebreton J.P. et al.: Serum concentration of human alpha 2 HS glycoprotein during the inflammatory process: evidence that alpha 2 HS glycoprotein is a negative acute-phase reactant. J Clin Invest. 1979, 64: 1118-1129.
49. Ketteler M. et al.: Association of low fetuin-A (AHSG) concentrations in serum with cardiovascular mortality in patients on dialysis:a cross-sectional study. Lancet. 2003, 361: 827-833.
50. Binkert C. et al.: Regulation of osteogenesis by fetuin. J Biol Chem. 1999, 274: 28514-28520.
51. Slatopolsky E. et al.: Calcium carbonate as a phosphate binder in patients with chronic renal failure undergoing dialysis. N. Engl. J. Med. 1986, 315: 157-161.
52. Chertow G.M. et al.: Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002, 62: 245-252.
53. Cannata-Andia J.B., Fernandez-Martin J.L.: The clinical impact of aluminium overload in renal failure. Nephrol. Dial. Transplant. 2002, 17 (Suppl. 2): 9-12.
54. Guillot A.P. et al.: The use of magnesium-containing phosphate binders in patients with end-stage renal disease on maintenance hemodialysis. Nephron 1982, 30: 114-117.
55. Chertow G.M. et al.: Long-term effects of sevelamer hydrochloride on the calcium x phosphate product and lipid profile of haemodialysis patients. Nephrol. Dial. Transplant. 1999, 14: 2907-2914.
56. D´Haese P.C. et al.: A multicenter study on the effects of lanthanum carbonate (Fosrenol) and calcium carbonate on renal bone disease in dialysis patients. Kidney Int 2003; 85 (Suppl.): S73-78.
57. Joy M.S., Finn W.F.: LAM-302 Study Group. Randomized, double-blind, placebo-controlled, dose-titration, phase III study assessing the efficacy and tolerability of lanthanum carbonate: a new phosphate binder for the treatment of hyperphosphatemia. Am J Kidney Dis. 2003, 42: 96-107.
58. Harrison T. S., Scott L. J.: Lanthanum carbonate. Drugs 2004, 64, 985-996.
59. 75. Block G.A. et al.: Cinalcet for secondary hyperparathyroidism in patients receiving hemodialysis. N Engl J Med 2004, 350, 1516-1525.
60. Mucsi I. et al.: Control of serum phosphate without any phosphate binders in patients treated with nocturnal hemodialysis. Kidney Int. 1998, 53: 1399-1404.