© Borgis - Postępy Nauk Medycznych 5/2008, s. 297-303
Joanna Skubis-Zegadło, Barbara Górka, Ewa Kubik, *Barbara Czarnocka
Neuronalna cząsteczka adhezyjna L1: struktura molekularna, funkcje biologiczne i rola w inwazji nowotworów
The neural cell adhesion molecule L1: molecular structure, biological functions and its role in the tumor invasion
Zakład Biochemii i Biologii Molekularnej, Centrum Medyczne Kształcenia Podyplomowego w Warszawie
Kierownik Zakładu: prof. dr hab. Andrzej Gardas
Streszczenie
W pracy przedstawiono postęp badań nad cząsteczką adhezyjną L1CAM. Cząsteczka ta w ostatnich latach stała się interesującym obiektem badań nie tylko w biologii, ale także w medycynie klinicznej. Omówione zostały aspekty budowy molekularnej L1CAM. Przedstawiono charakterystykę tego białka pełniącego ważne funkcje w oddziaływaniach międzykomórkowych. Opisano miejsca występowania L1CAM oraz jego ekspresję w warunkach fizjologicznych i patologii, głównie w nowotworach złośliwych. Opisano również rolę L1CAM jako potencjalnego markera nowotworowego. Ponadto zwrócono uwagę na mutacje w genie L1CAM i wynikające z tego zespoły chorobowe układu nerwowego.
Summary
Advances in the research on L1CAM: L1 cell adhesion molecule are presented. Recently L1CAM appears to become a very interesting molecule not only in biology, but also in clinical medicine. The aspects of molecular structure of L1CAM and its role in cell-cell interaction has been described. Main sites of L1CAM expression both in physiology and pathology, especially in malignancy, have been indicated. The L1 molecule function as a promising new biomarker for the diagnosis and prognosis of some human tumors has been also described. Moreover, the attention was paid to mutations in L1CAM gene which cause neurological disorders.

WSTĘP
Wzajemne oddziaływania komórek odgrywają ważną rolę w rozwoju organizmu, zwłaszcza w utrzymaniu integralności tkanek, w procesach naprawczych czy odpowiedzi immunologicznej. Właściwości adhezyjne komórek są związane z ekspresją różnych białek, które są odpowiedzialne za międzykomórkowe oddziaływania homo- i heterofilne. Należą do nich; integryny, kadheryny, selektyny oraz cząsteczki adhezyjne CAMs (cell adhesion molecules) należące do nadrodziny białek immunoglobulinopodobnych.
W procesie ewolucji, w wyniku duplikacji „przodka” genu L1 występującego u stawonogów, powstały u kręgowców 4 homologiczne typy genów L1 (1). U kręgowców występują cztery immunoglobulino podobne adhezyny: L1, neurofascyna, NrCAM (neuronalna cząsteczka adhezyjna) i CHL1 (close homolog of L1 – bliski homolog L1), podczas gdy u bezkręgowców dwa: neuroglian i traktyna (2). Homologi L1 odkryto również u gryzoni, ptaków, ryb oraz Drosophila (3).
Struktura molekularna L1
L1 jest błonową glikoproteiną zbudowaną z 1256 aminokwasów. W jej budowie wyróżnia się duży fragment zewnątrzkomórkowy zbudowany z 6 domen Ig-podobnych (Ig) związanych z 5 domenami fibronektyny typu III (Fn III), pojedynczy fragment przezbłonowy i krótki, filogenetycznie konserwowany fragment wewnątrzkomórkowy (ryc. 1) (3, 4). Budowa trzeciorzędowa L1 nie została dotychczas poznana. Przypuszcza się, że pierwsze cztery domeny Ig-podobne tworzą strukturę w kształcie podkowy, podobnie jak w hemolinie i aksoninie-1. Białka rodziny różnią się sekwencją i strukturą, ale większość z nich jest przypisana do jednego z czterech typów strukturalnych. W obrębie podrodziny L1 wyróżnia się 4 typy: V, C1, C2 i ostatni zidentyfikowany - I. Najbardziej znaną strukturę charakteryzującą typ I zbliżoną do struktury Ig-podobnych domen L1 posiada telokina. Wzory sekwencji aminokwasowych w ludzkim białku L1 wskazują jednoznacznie, że domeny od siódmej do jedenastej należą do nadrodziny fibronektyny typu III (Fn III). Cytoplazmatyczna domena L1 składa się z 85-148 aminokwasów (1). Domena ta jest bardzo silnie konserwowana ewolucyjnie, co pozwala sądzić, że spełnia ona ważne funkcje. Przypuszcza się również, że zmiany domeny cytoplazmatycznej L1 mogą wpływać na właściwości adhezyjne zewnątrzkomórkowych części cząsteczki białka. Wykazano, że cytoplazmatyczna domena L1 nie jest wymagana w interakcjach homofilnych (5,6).
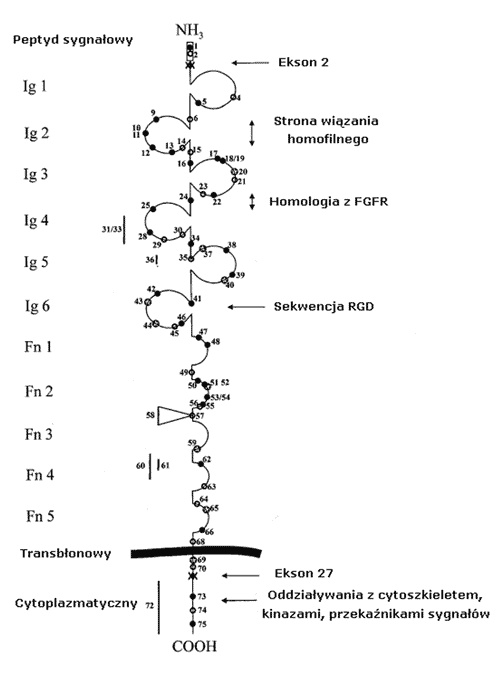
Ryc. 1. Schemat przedstawiający strukturę L1 oraz miejsca występowania mutacji wg Fransen i wsp. The
L1 protein and mutations in CRASH syndrome.
WYSTĘPOWANIE I FUNKCJE BIOLOGICZNE L1
Białko L1 i białka L1-podobne zidentyfikowano u ludzi, gryzoni, ryb, pierścienic.U ssaków L1 występuje w całym układzie nerwowym, zarówno w rozwijających i różnicujących się neuronach, jak i komórkach Schwanna. W różnicujących się neuronach L1 znajduje się w okolicach połączeń aksonalnych i w ich stożkach wzrostu. Adhezyjne właściwości L1 wpływają na tworzenie włókien nerwów osiowych. L1 może być zaangażowany we wzrost aksonów w trakcie rozwoju układu nerwowego, w oddziaływanie między aksonami a komórkami Schwanna, w migracje komórek neuronalnych, w synaptogenezę i mielinizację a nawet w przeżycie komórek nerwowych. Początkowo L1 odnajdowano wyłącznie w komórkach układu nerwowego. Badania ostatnich lat wykazały, że białko L1 znajduje się w szeregu typach komórek i tkanek, w tym tkanek nowotworowych (4, 8, 9). Ekspresja L1 w trakcie rozwoju kory mózgowej jest regulowana przez hormony tarczycy. Nieprawidłowa ekspresja L1 wynikająca z niedoczynności tarczycy może zostać naprawiona poprzez leczenie hormonalne (10).
Białka L1 wykazują homofilne interakcje z innymi cząsteczkami L1 występującymi na powierzchni przylegającej komórki a miejsce tego wiązania zlokalizowano na domenie Ig2. Wykazano, że w interakcjach heterofilnych z ektodomeną L1 reaguje szereg białek w tym: integryny, białka siateczki zewnątrzkomórkowej (laminina) i szereg proteoglikanów, w tym neurokan, fosfakan (ryc. 2) (3).
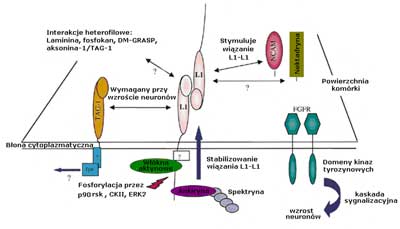
Ryc. 2. Schemat przedstawiający złożoność interakcji L1 wg Kenwrick i wsp. The complexity of L1 interaction.
Fragmenty zawierające sekwencję Arg-Gly-Asp (RGD) w szóstej domenie Ig-podobnej są miejscami receptorowymi integryn α5β1 (receptor fibronektyny), αvβ1, αvβ3 (receptor witronektyny). Poza fragmentem Ig-podobnym wykazano istnienie sekwencji RDG wiążącej integryny αvβ1 w trzecim segmencie domeny fibrynopodobnej. Tak więc, białko L1 spełniając funkcje liganda i receptora sprzyja adhezji komórek i wzrostowi neurytów w izolowanych neuronach (11).
Nie wszystkie interakcje L1 wywołują adhezję lub wzrost aksonu. L1 wiąże się również z powierzchniowymi proteoglikanami macierzy zewnątrzkomórkowej, czego efektem jest silne zahamowanie adhezji neuronalej i przerastania neurytu (4).
Istnieją dowody na to, że L1 może brać udział w wędrówce prekursorów neuronalnych. Lokalizacja prekursorów w „znokautowanych” myszach różni się od lokalizacji u osobników żyjących w warunkach naturalnych. L1 stymuluje rozrost neuronów w hodowlach komórkowych. Wykorzystuje do tego mechanizm centralnego integratora sygnałów: kinazę MAP (12). Wykazano, że L1 wspomaga indukowaną integryną αvβ3 migrację komórek. Kompleks L1- integryna αvβ3 odgrywa istotną rolę w procesach migracji leukocytów (13).
SYGNALIZACJA
Wpływ, jaki wywiera L1 na zachowanie innych komórek wskazuje, że białko to jest czynnikiem związanym z wewnątrzkomórkowymi szlakami sygnalizacyjnymi. W jaki sposób glikoproteina występująca na powierzchni komórki, nieposiadająca domen katalitycznych może być zaangażowana w sygnalizację? Odbywa się to poprzez wiązanie L1 do cząsteczek, które same są zdolne wywołać sygnał. Stymulowanie wzrostu aksonów spowodowane jest aktywowaniem związanych z kinazami tyrozynowymi receptorów czynników wzrostu fibroblastów (4).
Fosforylacja jest powszechnie występującym mechanizmem regulującym wewnątrzkomórkowe kaskady sygnalizacyjne. L1 ulega glikozylacji i fosforylacji. Zidentyfikowanie miejsc fosforylacji L1 i kinaz odpowiedzialnych za fosforylację L1 może pozwolić wyjaśnić mechanizm wpływu L1 na rozwój neuronów. Zidentyfikowano trzy kinazy biorące udział w fosforylacji L1: CKII, p90rsk, ERK2, które in vitro fosforylują niektóre aminokwasy serynowe w domenie cytoplazmatycznej cząsteczki L1.
Wykazano, że konstytutywna aktywacja kinazy ERK (składnik ścieżki kinazy MAP) ma istotne znaczenie w procesie transformacji komórkowej i rozwoju różnych nowotworów. Kinaza ERK jest aktywowana przez L1. Wykazano, że ekspresja L1 u myszy powoduje zwiększenie zdolności rakotwórczych komórek. W przypadku nowotworu okrężnicy u ludzi również zaobserwowano, że indukcja ekspresji L1 w hodowlach komórek pozbawionych genu L1 prowadzi do zwiększenia właściwości inwazyjnych i przyspieszenia wzrostu nowotworu. Wygaszanie endogennego L1 przez siRNA w komórkach nowotworowych okrężnicy obniżało wspomniane wyżej zdolności. Wykazano, że neoekspresja L1 w komórkach NIH3T3 (mysie fibroblasty embrionalne) powoduje nabieranie przez nie pewnych cech komórek nowotworowych takich jak zwiększona ruchliwość i transformacja komórek. Stąd wydaje się, białka z rodziny L1 mogą być potencjalnymi promotorami rozwoju i inwazji komórek raka okrężnicy (14).
MUTACJE W GENIE L1CAM I ZWIĄZANE Z TYM CHOROBY UKŁADU NERWOWEGO
W 1949 Bickers i Adams opisali przypadek brytyjskiej rodziny, w której u większości mężczyzn przyczyną zgonu było wrodzone wodogłowie. Badania pośmiertne wykazały zniekształcenia strukturalne mózgu: zwężający się wodociąg śródmózgowia. Występowanie wodogłowia wywołane jest najprawdopodobniej zwężeniem wodociągu. Zespół ten nazwano „wodogłowiem wywołanym zwężeniem wodociągu śródmózgowia, lub wodociągu Sylwiusza” (HSAS – Hydrocephalus due to Stenosis of the Acqueduct of Sylvius). Kliniczne i patologiczne cechy tej recesywnej, sprzężonej z płcią choroby poznano bardzo dokładnie. Częstość występowania tego zespołu ocenia się średnio 1 na 30 000 narodzin męskich osobników. W większości przypadków u pacjentów z wodogłowiem sprzężonym z płcią obserwuje się bardzo zróżnicowane nieprawidłowości. Jedyną wspólną cechą HSAS jest niedorozwój umysłowy ze wskaźnikiem IQ w zakresie od 20 do 50. Większość chorych cierpi na spastyczną paraplegię, co w większości wypadków jest spowodowane zwyrodnieniem dróg korowo-rdzeniowych. Ponadto odnotowywane są również przypadki agenezji lub dysgenezji ciała modzelowatego lub przegrody przezroczystej (3).
U ludzi gen L1jest zlokalizowany w pobliżu telomeru na długim ramieniu chromosomu X (Xq28). Gen kodujący L1 znajduje się pomiędzy locus ALD i MeCP2. Wielkość genu L1 od miejsca startu translacji do kodonu stop wynosi 12942 bp a gen zbudowany jest z 28 eksonów. Alternatywne składanie (mechanizm konserwowany od ponad 430 milionów lat) specyficzne dla różnych tkanek prowadzi do ominięcia eksonów 2 i 27, skutkiem czego powstają różne izoformy L1. Na przykład w mięśniach nie odnajduje się transkryptów eksonu 2 i 27, podczas gdy są one obserwowane w mózgu. Struktura genowa L1jest silnie konserwowana, szczególnie wśród ssaków. Zachowana jest u nich bardzo duża identyczność aminokwasowa (80-95%) w domenach zewnątrzkomórkowych i całkowita zgodność aminokwasowa w domenie cytoplazmatycznej (15,16).
Mutacje w obrębie genu L1dotyczą zarówno domen zewnątrz- jak i wewnątrzkomórkowych. Mutacje, które występują w obrębie genu L1 można podzielić na dwie grupy: mutacje eliminujące zdolności zakotwiczenia na powierzchni komórki (mutacje braku sensu i mutacje zmiany ramki odczytu regionu zewnątrzkomórkowego) i mutacje, które wywierają subtelniejszy efekt na strukturę i funkcje białka (mutacje zmiany sensu). W obrębie mutacji zmiany sensu wyróżnia się mutacje dotyczące domen zewnątrzkomórkowych i domen wewnątrzkomórkowych (17).
Mutacje w genie kodującym L1wywo-łują szerokie spektrum wad układu ner-wowego. Zidentyfikowano 85 różnych mutacji genu L1. Mutacje zmiany sensu mogą wpływać na funkcjonowanie białka na kilka różnych sposobów. Jeśli spowodują nieprawidłowe fałdowanie białka, przeszkodzi to w prezentacji specyficznego miejsca wiązania liganda lub rozerwie czwartorzędową strukturę białka. Mutacje zmiany sensu, które nie niszczą struktury drugorzędowej mogą wpływać na interakcje wewnątrz domenowe lub uszkadzać miejsca wiązania białko - białko (18).
U osobników z mutacjami w genie L1diagnozowano: XLH (X – linked hydrocephalus), zespół MASA (Mental retardation, Aphasia, Spastic paraplegia and Adducted thumbs), sprzężony z płcią niedowład spastyczny lub sprzężoną z płcią agenezję ciała modzelowatego (19). Większość z tych fenotypów obserwowano u osobników jednej rodziny. Pozwoliło to wysunąć hipotezę, że choroby te są wynikiem plejotropowych efektów mutacji w jednym genie. Zaprzestanie traktowania oddzielnie chorób związanych z mutacjami w L1przyczyniło się do powstania nowego terminu: syndromu CRASH (Corpus callosum agenesis, Retardation, Adducted thumbs, Spastic paraplegia and Hydrocephalus). Nazwa ta odnosi się do wszystkich klinicznych objawów mutacji w genie kodującym L1. Mutacje w tym genie mogą się prawdopodobnie przyczynić do powstania zespołu MRX3 i PH. MRX3 jest formą specyficznego niedorozwoju umysłowego związanego z chromo-somem Xq28. Okołokomorowa heterotopia (PH – Periventricular Heterotopia) jest wywołana defektem migracyjnym w trakcie rozwoju mózgowo-korowego. PH jest często związana z występowaniem ogniskowych padaczek lekoopornych. W obu przypadkach zaproponowano, że odpowiedzialne są za to mutacje w genie L1, ale nie udowodniono jeszcze tego założenia (4, 20).
L1CAM I NOWOTWORY
Onkogeneza jest wielostopniowym procesem związanym ze zmianami genetycznymi obdarzającymi komórki nowotworowe po pierwsze zwiększonymi właściwościami proliferacyjnymi, a w zaawansowanych stadiach zdolnością do rozluźnienia adhezji między-komórkowej i nabycia zdolności poruszania się. Pozwala to komórkom nowotworowym na naciekanie sąsiednich tkanek i tworzenie odległych przerzutów. Powyższe procesy związane są ze zmianami w ekspresji białek i w procesach sygnalizacyjnych odpowiedzialnych za adhezję komórkową. Nadekspresja L1 w wielu nowotworach w porównaniu do tkanek prawidłowych zapoczątkowała szerokie badania nad jej znaczeniem w biologii tych guzów, ale także badania nad możliwością oznaczania białka L1, jako markera nowotworowego. Jak się wydaje, cząsteczka adhezyjna L1 jest obiecującym markerem nowotworowym. Dobry biomarker, powinien pozwalać określić występowanie specyficznego nowotworu z dużą dokładnością i mógłby zostać użyty nie tylko do rozpoznawania, określenia stopnia zaawansowania, ale również do monitorowania skuteczności chemio- lub radioterapii i wykrywania wznowy guza.
Cząsteczka adhezyjna L1 jak wykazały badania in vitro jest związana z mechanizmami kontrolującymi proliferację, migrację i procesy inwazji szeregu nowotworów. Pierwsze prace donosiły o ekspresji i funkcji L1 w komórkach nowotworowych pochodzących z płuc, nerek i skóry, białaczkach monocytowych i mięsaku mięśni poprzecznie prążkowanych oraz w wielu nowotworowych liniach komórkowych uzyskanych od ludzi i gryzoni jak linia neuroektodermalna (czerniak i nerwiak zarodkowy współczulny). Badania nad identyfikacją onkogenów wykazały, że jednym z kandydatów w rakach jajnika może być gen kodujący białko adhezji komórkowej L1. Wykazano wysoką ekspresję L1 w rakach wywodzących się z komórek nabłonka i bardzo rzadko w innych typach raków jajnika. Zastosowanie przeciwciał monoklonalnych L1-11A przeciwko ludzkiemu L1 pozwoliło na bardzo szerokie badania nad przydatnością oznaczania tego białka metodą immunohistochemiczną dla diagnozowania bardzo agresywnych raków u ludzi jak raka jajnika, endometrium czy rak jelita grubego (21, 22).
Cząsteczka adhezyjna L1 może słu-żyć jako marker w diagnozowaniu no-wotworów jajnika i macicy. Nowotwór endometrium jest jednym z najczęściej występujących i diagnozowanych nowotworów w krajach wysoko rozwiniętych. W 2001 roku w USA zanotowano 38 300 zachorowań na raka macicy, w tym 6600 przypadków śmiertelnych.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Hortsch M: Structural and functional evolution of the L1 family: are four adhesion molecules better than one? Mol Cel Neurosci 2000; 15: 1-10.
2. Brümmendorf T, et al.: Neural cell recognition molecule L1: from cell biology to human hereditary brain malformations. Curr Opin Neurobiol 1998; 8: 87-97.
3. Fransen E, et al.: L1-associated diseases: clinical geneticists divide, molecular geneticists unite. Hum Mol Genet 1997; 6: 1625--1632.
4. Kenwrick S, et al.: Neural cell recognition molecule L1: relating biological complexity to human disease mutations. Hum Mol Genet 2000; 9: 879-886.
5. Sheafer AW, et al.: L1 endocytosis is controlled by a phosphorylation-dephosphorylation cycle stimulated by outside - in signaling by L1. J Cell Biol 2002; 157: 1223-1232.
6. Wong E, et al.: The cytoplasmic domain of the cell adhesion molecule L1 is not required for homophilic adhesion. Neurosci Lett 1995; 200: 155-158.
7. Gutwein P, et al.: Role of SRC kinases in the ADAM-mediated release of L1 adhesion molecule from human tumor cells. J Biol Chem 2000; 275: 15490-15497.
8. Kadmon G, et al.: Functional cooperation between the neural adhesion molecules L1 and N-CAM is carbohydrate dependent. J Cell Biol 1998; 110: 209-218.
9. Thelen K, et al.: The neural cell adhesion molecule L1 potentiates integrin - dependent cell migration to extracellular matrix proteins. J Neuroscience 2002; 22: 4918-4931.
10. Dolado-Alvarez M, et al.: Regulation of the L1 cell adhesion molecule by thyroid hormone in the developing brain. Mol Cell Neuroscience 2000; 16: 499-514.
11. Mechtersheimer S, et al.: Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J Cell Biol 2001; 155: 661-673.
12. Shaefer AW, et al.: Activation of the MAPK signal cascade by the neural cell adhesion molecule requires L1 internalization. J Biol Chem 1999; 274: 37965-37973.
13. Duczmal A, et al.: The L1 adhesion molecule supports αvβ3-mediated migration of human tumor cells and activated T lymphocytes. Biochem Biophys Res Com 1997; 232: 236-239.
14. Gavert N, et al.: L1, a novel target of β-catenin signaling, transforms cells and is expressed at the invasive front of colon cancers. J Cell Biol 2005; 168: 633-642.
15. Coutelle O, et al.: The neural cell adhesion molecule L1: genomic organisation and differential splicing is conserved between man and the pufferfish Fugu. Gene 1998; 208: 7-15.
16. Miura M, et al.: Molecular cloning of cDNA encoding the rat neural cell adhesion molecule L1. Two isoforms in the cytoplasmic region are produced by differential splicing. FEBS Lett 1991; 289: 91-95.
17. Bateman A, et al.: Outline structure of the human L1 cell adhesion molecule and the sites where mutations cause neurological disorders. EMBO J 1996; 15: 6050-6059.
18. De Angelis E, et al.: Disease-associated mutations in L1 CAM interfere with ligand interactions and cell surface expression. Hum Mol Genet 2002; 11: 1-12.
19. Kamiguchi H, et al.: Role of L1 in neural development: what the knockouts tell us. Mol Cell Neurosci 1998; 12: 48-55.
20. De Angelis E, et al.: Pathological missense mutations of neural cell adhesion molecule L1 affects homophilic and heterophilic binding activities. EMBO J 1999; 18: 4744-4753.
21. Fogel M, et al.: L1 (CD171) as a novel biomarker for ovarian and endometrial carcinomas. Expert Rev Mol Diagn 2004; 4: 455-462.
22. Deichmann M, et al.: Adhesion molecules CD171 (L1CAM) and CD24 are expressed by primary neuroendocrine carcinomas of the skin (Merkel Cell carcinomas). J Cutan Pathol 2003; 30: 363-368.
23. Euer NI, et al.: Identification of L1CAM, Jagged2 and Neuromedin U as ovarian cancer-associated antigens. Oncol Rep 2005; 13: 375-387.
24. Fogel M, et al.: L1 expression as a predictor of progression and survival in patients with uterine and ovarian carcinomas. Lancet 2003; 362: 869-875.
25. Fogel M, et al.: L1 adhesion molecule (CD171) in development and progression of human malignant melanoma. Cancer Lett 2003; 189: 237-247.
26. Kettunen E, et al.: L1CAM, INP10, P-cadherin, tPA and ITGB4 over-expression in malignant pleural mesotheliomas revealed by combined use of cDNA and tissue microarray. Carcinogenesis 2005; 26: 17-25.
27. Suzuki T, et al.: Clinicopathological study of cellular proliferation and invasion in gliomatosis cerebri: important role of neural cell adhesion molecule L1 in tumour invasion. J Clin Pathol 2005; 58: 166-171.
28. Debiec H, et al.: The cell adhesion molecule L1 developmentally regulated in the renal epithelium and is involved in kidney branching morphogenesis. J Cell Biol 1998; 143: 2067-2079.
29. Allory Y, et al.: The L1 cell adhesion molecule is induced in renal cancer cells and correlates with metastasis in clear cell carcinomas. Clin Cancer Res 2005; 11: 1190-1197.
30. Primiano T, et al.: Identification of potential anticancer drug targets through the selection of growth-inhibitory genetic supressor elements. Cancer Cell 2003; 4: 41-53.
31. Gelman MS, et al.: Identification of cell surface and secreted proteins essential for tumor cell survival using a genetic suppressor element screen. Oncogene. 2004; 23: 8158-8170.
