Genetyka kliniczna raka piersi i jajnika
Hereditary breast and ovarian cancer
1 Międzynarodowe Centrum Nowotworów Dziedzicznych Zakład Genetyki i Patomorfologii Pomorska Akademia Medyczna, Szczecin
Kierownik Zakładu Genetyki i Patomorfologii: prof. dr hab. med. Jan Lubiński
2 Katedra i Klinika Ginekologii Operacyjnej i Onkologii Ginekologicznej Dorosłych i Dziewcząt Pomorska Akademia Medyczna, Szczecin
Kierownik: prof. dr hab. med. Izabella Rzepka-Górska
Streszczenie
W ostatnich latach udało się wykazać u niemal wszystkich pacjentów z rakami piersi lub jajnika charakterystyczne podłoże konstytucyjne sprzyjające rozwojowi tych nowotworów. Stwierdzono, że nosicielstwo mutacji w genach BRCA1, BRCA2, CHEK2, NBS1, NOD2, CDKN2A, CYP1B1jak i rzadziej występujących zmian w genach takich jak ATM, PTEN, STK11wiąże się z podwyższonym ryzykiem raka piersi. Zaburzenia w genach BRCA1, BRCA2, NOD2, CHEK2, DHCR7predysponują do rozwoju raka jajnika. W niektórych przypadkach zmiany genetyczne wiążą się z bardzo wysokim ryzykiem nowotworowym, w innych przypadkach wykrywane zaburzenia predysponują do rozwoju raka w mniejszym stopniu. Zdiagnozowanie podwyższonego ryzyka raka umożliwia wdrożenie programu profilaktycznego umożliwiającego zapobieżenie nowotworowi, a tam gdzie to się nie udaje pozwala na wykrycie raka we wczesnym stadium. Dodatkowo zdiagnozowanie nosicielstwa odpowiednich mutacji pozwala na dobór najefektywniejszego, zindywidualizowanego sposobu leczenia związanego z uwarunkowaniami konstytucjonalnymi pacjenta. Dużym problemem diagnostycznym są pacjentki, u których zmian molekularnych nie udało się znaleźć, ale dane rodowodowo-kliniczne wskazują na silne podłoże genetyczne nowotworu. W niniejszym opracowaniu przedstawiono podłoże genetyczne rozwoju raka piersi i jajnika uwzględniając wpływ genów wysokiego oraz umiarkowanie zwiększonego ryzyka jak i zasady interpretacji danych rodowodowych. Omówiono obecnie obowiązujące zasady diagnozowania grup ryzyka, profilaktyki oraz leczenia raka u pacjentek z zespołem BRCA1, BRCA2, BRCAX oraz ze zmianami w genach umiarkowanie zwiększonego ryzyka.
Summary
Recently, it is possible to show a constitutional genetic background in almost all patients with breast or ovarian cancer. This was shown in carriers of mutations in BRCA1, BRCA2, CHEK2, NBS1, NOD2, CDKN2A, CYP1B1 and less frequently of genes such as ATM, PTEN, STK11. Abnormalities in BRCA1, BRCA2, NOD2, CHEK2, DHCR7 genes are predisposing factors also for development of ovarian cancer. In some cases, characteristic gene mutations are related to a very high risk of cancer, in other cases detected genetic changes predispose to cancer at lower degree. Diagnosis of increased risk of cancer allows introduction of prophylactic programs which make possible to avoid cancer, or diagnose it in early stages as well as application of most effective method of treatment individualized for specific mutation carrier. Significant diagnostic problem constitute patients where molecular abnormality was not detected but pedigree-clinical data indicate strong genetic background of cancer. In the review we show the genetic background of breast and ovarian cancer taking into consideration contribution of high and moderate penetrance genes as well as importance of pedigree data. We discuss rules of diagnosis, prophylactics, the most sensitive methods of early detection and treatment in patients with BRCA1, BRCA2 and BRCAX syndromes as well as in patients with abnormalities in moderate penetrance genes.

Najstarsze doniesienie o rodzinnym raku piersi datuje się na około 100 rok naszej ery i pochodzi z literatury medycznej Starożytnego Rzymu (1). Pierwsza dokumentacja rodzinnej agregacji raka piersi pochodząca z czasów nowożytnych została opublikowana przez Broca w 1866 roku, który opisał 10 przypadków raka piersi w 4 pokoleniach rodziny swojej żony (2). W połowie lat dziewięćdziesiątych udowodniono również na poziomie molekularnym, że znacząca część raków piersi i jajnika ma dziedziczną etiologię jednogenową (3, 4). Badania oceniające częstość występowania cech rodowodowo-klinicznych charakterystycznych dla silnych agregacji raków piersi/jajnika wśród kolejnych raków tych narządów jak i analizy zgodności zachorowań wśród bliźniaków jednojajowych wskazują, że w około 30% raków piersi i jajnika zachorowania te powstają wskutek silnej genetycznej predyspozycji (5). Do niedawna w pozostałych tzw. sporadycznych rakach piersi/jajnika znaczenie czynników genetycznych było pomijane. W ostatnich latach udało się jednak wykazać, że u pacjentów z rakami sporadycznymi również jest wykrywalne charakterystyczne podłoże konstytucyjne sprzyjające rozwojowi tych nowotworów. Dlatego obecnie uważa się, że u niemal wszystkich pacjentów z nowotworami powinno występować odpowiednie podłoże genetyczne, oczywiście w różnym stopniu wpływające na ryzyko rozwoju nowotworu. Zmiany genetyczne silnie związane z występowaniem nowotworu określa się jako zmiany (geny) wysokiego ryzyka, natomiast zmiany powiązane z rozwojem danego nowotworu w mniejszym stopniu nazywa się zmianami (genami) umiarkowanie zwiekszonego ryzyka. Klinicznie silna genetyczna predyspozycja do raka piersi/jajnika jest na ogół powiązana z mutacjami w genach BRCA1lub BRCA2i ujawnia się najczęściej jako zespoły tzw. dziedzicznego raka piersi specyficznego narządowo (hereditary breast cancer – site specific; HBC-ss), dziedzicznego raka piersi-jajnika (hereditary breast-ovarian cancer; HBOC) i dziedzicznego raka jajnika specyficznego narządowo (hereditary ovarian cancer; HOC). W zespole HBC-ss u członków rodzin występują raki piersi a nie stwierdza się raków jajnika, w zespole HBOC wśród krewnych rozpoznawane są zarówno raki piersi jak i jajnika, w zespole HOC w rodzinach występują raki jajnika natomiast nie stwierdza się raków piersi. Na podstawie cech rodowodowo-klinicznych charakterystycznych dla raków piersi/jajnika związanych z mutacjami o wysokiej penetracji określono kryteria umożliwiające rozpoznawanie definitywne lub z wysokim prawdopodobieństwem rodzin z zespołami HBC-ss, HBOC, HOC. Kryteria te zestawiono w tabeli 1. W zdecydowanej większości przypadków nowotworów związanych z genami umiarkowanie zwiekszonego ryzyka wywiad rodzinny jest nieobciążony.
Tabela 1. Kryteria rodowodowo-kliniczne rozpoznawania zespołów HBC-ss, HBOC i HOC (6).
| Liczba przypadków raka piersi lub jajnika w rodzinie: |
| A - trzy (diagnoza definitywna) |
| 1. Przynajmniej 3 krewnych dotkniętych rakiem piersi/jajnika rozpoznanym w dowolnym wieku; |
| B - dwa (diagnoza z dużym prawdopodobieństwem) |
| 1. 2 raki piersi lub jajnika wśród krewnych Io (lub IIo przez mężczyznę); |
| 2. 1 rak piersi i 1 rak jajnika rozpoznane w dowolnym wieku wśród krewnych Io (lub IIo przez mężczyznę); |
| C - jeden (diagnoza z dużym prawdopodobieństwem) |
| 1. Wystąpienie raka piersi poniżej 40 roku życia; |
| 2. Wystąpienie raka piersi obustronnego; |
| 3. Wystąpienie raka piersi rdzeniastego lub atypowego rdzeniastego; |
| 4. Wystąpienie raka piersi i jajnika u tej samej osoby; |
| 5. Wystąpienie raka piersi u mężczyzny; |
Zespoły HBC-ss, HBOC, HOC są heterogenne klinicznie i molekularnie. Do najczęstszych przyczyn ich powstawania należą mutacje konstytucyjne w genach BRCA1i BRCA2.
Zespół BRCA1
W zespole tym stwierdza się u pacjentki konstytucyjną mutację genu BRCA1. U nosicielek mutacji tego genu obserwuje się około 50-80% ryzyko rozwoju raka piersi i około 40% ryzyko rozwoju raka jajnika (7). Wg danych uzyskanych w naszym Ośrodku dla populacji polskiej na podstawie badania kolejnych raków piersi/jajnika powyższe wielkości wynoszą odpowiednio około 66% dla raka sutka i 44% dla raka jajnika (tab. 2). W ostatnim czasie zaobserwowano, że ryzyko jest uzależnione od rodzaju mutacji i lokalizacji w genie (8, 9, 10). Wg naszych obserwacji np. ryzyko zachorowania na raka piersi jest około 2-krotnie wyższe u nosicielek 5382insC, w porównaniu z ryzykiem u nosicielek 4153delA.
Tabela 2. Ryzyko raka piersi i jajnika u nosicielek mutacji BRCA1w Polsce (8).
| A: Skumulowane ryzyko raka piersi: |
| Wiek: | <30 | 40 | 50 | 60 | 70 | 75 |
| Ryzyko skumulowane (%): | 1,6 | 6,5 | 30 | 40,5 | 50,5 | 66 |
| B: Skumulowane ryzyko raka jajnika: |
| Wiek: | <30 | 40 | 50 | 60 | 70 | 75 |
| Ryzyko skumulowane (%): | 1 | 3,5 | 12 | 30 | 41 | 44 |
Niepełna penetracja BRCA1sugeruje, że inne genetyczne i pozagenetyczne czynniki mają znaczenie w karcinogenezie u nosicieli mutacji. Opisano, że ryzyko rozwoju raka jajnika jest modyfikowane przez VNTR lokus dla HRAS 1 – ryzyko raka jajnika jest 2-krotnie większe dla nosicieli mutacji BRCA1posiadających jeden lub dwa rzadkie allele HRAS 1 (11). Badania przeprowadzone w naszym Ośrodku wśród kobiet z mutacją genu BRCA1wykazały, że obecność polimorfizmu 135G>C w genie RAD51 związane jest z dwukrotnym zmniejszeniem ryzyka zachorowania na raka sutka oraz jajnika (12, 13).
Charakterystyczne dla raków jajnika u nosicielek mutacji BRCA1jest również zwiększone ryzyko raków jajowodu i otrzewnej szacowane na około 10% (14). Przedstawione powyżej dane o częstości zachorowań na raka jajnika dotyczą najprawdopodobniej częstości raków jajnika, jajowodu i otrzewnej łącznie, ponieważ te ostatnie guzy były w przeszłości najczęściej rozpoznawane jako raki jajnika ze względu na podobieństwo obrazu histologicznego i towarzyszący im wzrost poziomu markera CA 125.
Najprawdopodobniej ryzyko raków innych narządów w niektórych rodzajach mutacji BRCA1jest również zwiększone, jednak ten efekt nosicielstwa zaburzeń BRCA1nie został dotąd dowiedziony ostatecznie.
Raki piersi i jajnika zależne od BRCA1wykazują szereg charakterystycznych cech klinicznych. Średni wiek diagnozowania raków piersi tego typu wynosi około 42-45 lat (15, 16) a raków jajnika około 54 lat (17, 18). Obustronność stwierdza się w około 18-32% raków piersi BRCA1zależnych (19, 20). Bardzo charakterystyczną cechą jest szybkie tempo rozrastania się tych guzów – w ponad 90% przypadków raki BRCA1zależne wykazują G3 – trzeci stopień morfologicznej złośliwości już w chwili rozpoznania. Niemal wszystkie raki jajnika u nosicielek mutacji BRCA1diagnozowane są też w III°/IV° zaawansowania klinicznego wg FIGO. Raki piersi często są rdzeniaste, atypowe rdzeniaste lub przewodowe bez wykrywalnej obecności receptorów estrogenowych (ER). Raki piersi zależne od BRCA1stanowią około 10-15% wszystkich raków ER – (21, 22, 23). Wywiad rodzinny w większości przypadków jest obciążony (ryc. 1), jednak ze względu na dziedziczenie na linię ojcowską i niepełną penetrację przypadki z negatywnym wywiadem rodzinnym stanowią ok. 45% (ryc. 2) (20).
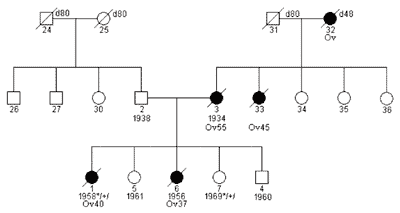
Ryc. 1. Rodzina z zespołem HOC oraz stwierdzoną mutacją konstytucyjną genu BRCA14153delA.
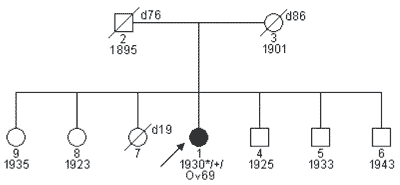
Ryc. 2. Osoba z rakiem jajnika i ze stwierdzoną mutacją konstytucyjną genu BRCA1– 5382insC z rodziny bez innych uchwytnych cech rodowodowo-klinicznych charakterystycznych dla rodzin z zespołem HBOC/HOC.
Diagnostyka molekularna mutacji konstytucyjnych genu BRCA1
Problem ten został opisany we wcześniejszym artykule – „Test BRCA1”.
Zespół BRCA2
W zespole tym stwierdza się u pacjentki konstytucyjną mutację genu BRCA2 (24). Na podstawie danych z piśmiennictwa w rodzinach z definitywnym HBC-ss i HBOC u nosicielki mutacji BRCA2ryzyko raka piersi sięga 31-56% a raka jajnika 11-27% (10, 25, 26, 27, 28). Jak wykazały badania 200 polskich rodzin z silną agregacją raków piersi/jajnika, mutacje konstytucyjne genu BRCA2występują w tej grupie rzadko – z częstością około 4%. Nie jest jak dotąd znane ryzyko skumulowane rozwoju raka u polskich nosicieli mutacji genu BRCA2. Większość mutacji BRCA2występujących w naszej populacji najprawdopodobniej na ogół nieznacznie zwiększa ryzyko raka piersi, chociaż badania wykonane w naszym Ośrodku wykazały, że w rodzinach z agregacją raka piersi zdiagnozowanego przed 50 r.ż. oraz żołądka zdiagnozowanego u mężczyzn przed 55 r.ż. częstość mutacji genu BRCA2 wystepuje na poziome 10-20% (29). Mutacje genu BRCA2 wiążą się natomiast ze znacznym, chociaż dokładniej nieokreślonym, ryzykiem raka jajnika oraz raków przewodu pokarmowego – żołądka, jelita grubego, trzustki i to zarówno u kobiet jak i u mężczyzn. Przemawiają za tym badania przeprowadzone w naszym Ośrodku, w których mutacje wykrywano z częstością około 30% w rodzinach bez raka piersi, ale z agregacją raka jajnika oraz raka żołądka, jelita grubego lub trzustki wśród krewnych I° lub II° niezależnie od płci (30). Z badań wykonanych w Poznaniu wynika, że częstość mutacji BRCA2jest też zwiększona w rodzinach z rakiem piersi u mężczyzn i wynosi ona w naszym kraju około 15% (31).
Raki piersi i jajnika w rodzinach z mutacjami BRCA2wykazują szereg cech charakterystycznych. Według danych z piśmiennictwa średni wiek zachorowania na raki zależne od BRCA2wynosi dla raków piersi 52 lata u kobiet i 53 lata u mężczyzn oraz dla raków jajnika 62 lata (31, 32).
Diagnostyka molekularna nosicielstwa mutacji BRCA2
W odróżnieniu od genu BRCA1, jak dotąd nie opisano dla naszej populacji znaczącego stopnia „efektu założyciela” dla mutacji genu BRCA2 (22). Jego istnienie wydaje się prawdopodobne w świetle innych wyników badań w Polsce nad grupą genów, w których względnie rzadko występują zaburzenia „de novo”. W związku z powyższym mutacje BRCA2należy wykrywać indywidualnie dla każdej rodziny, co wymaga sekwencjonowania. Koszt tego badania jest wysoki i wynosi około 5000 PLN ze względu na wielkość genu BRCA2– 70 kpz genomowego DNA.
W rodzinach ze znalezioną konstytucyjną mutacją markerową u probanta koszt analizy molekularnej z dwóch niezależnych pobrań krwi potwierdzającej lub wykluczającej nosicielstwo mutacji BRCA2dla każdego z pozostałych krewnych wynosi około 400 PLN.
Zespół BRCAX
W Polsce w około 30% rodzin z rozpoznanymi definitywnie zespołami HBC-ss i HBOC oraz w około 40% rodzin z zespołem HOC nie są wykrywane mutacje BRCA1lub BRCA2. W pojedynczych przypadkach tych rodzin można rozpoznać jeden z rzadkich zespołów zestawionych w tabeli 3, w przebiegu, których występują ze zwiększoną częstością raki piersi/jajnika. W wielu ośrodkach na świecie nadal trwają prace nad identyfikacją genów, których mutacje powodują BRCAX.
Tabela 3. Wybrane rzadkie zespoły genetyczne ze zwiększonym ryzykiem występowania raka piersi i/lub jajnika.
| Schorzenie | Obraz kliniczny | Mutacje genu / Dziedziczenie | Piśmiennictwo |
| Zespół Li-Fraumeni | raki piersi, mięsaki, guzy mózgu, białaczka, raki nadnercza | p53,wysoka penetracja; AD | 17, 33 |
| Choroba Cowdena | wieloogniskowe zaburzenia śluzowoskórne, łagodne choroby proliferacyjne różnych organów, raki tarczycy, raki piersi/jajnika | PTEN AD | 34, 35 |
| HNPCC | raki jelita grubego, trzonu macicy i innych organów włączając raka piersi/jajnika | MSH2, MLH1; AD | 36 |
| Zespół Peutz-Jeghers | śluzowoskórna pigmentacja melaninowa, polipy jelitowe, raki kolorektalne i jelita cienkiego, guzy gonadalne, rak piersi | STK11;AD | 37 |
| Zespół Ruvalcaba-Myhre-Smith (Z.Bannayan-Riley-Ruvalcaba) | makrocefalia, polipy jelitowe, plamy
"café-au-lait" na prąciu, tłuszczaki, raki tarczycy i piersi | PTEN AD | 38 |
| Heterozygotyczne nosicielstwo mutacji genu dla "ataxia telangiectasia" | ataksja móżdżkowa, telangiektazje oczne i skórne, nadwrażliwość na promieniowanie radiacyjne, różne nowotwory włączając raka piersi/jajnika | ATM | 39 |
| Nosiciele mutacji genu ATH | zwiększone ryzyko rozwoju raka piersi u kobiet | niska penetracja 20-40%; AD | 6 |
| Zespół Klinefeltera | gynecomatia, cryptorchidism, guzy z ekstragonadalnych komórek germinalnych germ cell tumors, rak piersi u mężczyzn | 47, XXY; niska penetracja < 10% | 40 |
| Mutacja genu receptora androgenowego | rodzinne raki piersi u mężczyzn | Receptor androgenowy | 41 |
| Konstytucjonalna translokacja t (11q;22q) | zwiększone ryzyko rozwoju raka piersi | translokacja zrównoważona t (11q;22q) | 42 |
Rodowodowe dziedziczenie:
AD - autosomalne dominujące
AR - autosomalne recesywne |
Zalecenia postępowania w rodzinach z wysokim ryzykiem raka piersi/jajnika
Specjalne zasady postępowania należy zastosować u:
– nosicieli mutacji genów związanych z wysokim ryzykiem raka piersi/jajnika, jeśli takie mutacje zostały wykryte w rodzinie; w takich przypadkach zwykle tylko około 50% członków rodziny musi być włączonych do programu
– wszystkich członków rodzin z rozpoznaniem definitywnym lub podejrzeniem zespołu dziedzicznego raka piersi/jajnika według kryteriów rodowodowo-klinicznych zestawionych w tabeli 1, jeśli konstytucyjne mutacje predysponujące do rozwoju raków nie zostały wykryte.
Specjalne postępowanie dotyczy:
a) profilaktyki;
b) schematu badań kontrolnych;
c) leczenia.
Profilaktyka
Doustna hormonalna antykoncepcja
Dobrze udokumentowano przeciwwskazania do stosowania doustnych środków antykoncepcyjnych przez nosicielki mutacji BRCA1w wieku do 25 lat.
Wykazano, że środki te stosowane w młodszym wieku przez 5 lat zwiększają ryzyko raka piersi nawet o 35% (43). Ponieważ w około 30% przypadków u nosicielek mutacji BRCA1nie stwierdza się jakichkolwiek cech HBC-ss, HBOC lub HOC, wydaje się konieczne wykonywanie testu BRCA1u każdej młodej kobiety, która decyduje się na doustną antykoncepcję. Środki antykoncepcyjne stosowane przez nosicielki mutacji BRCA1po 30 roku życia wydają się nie wpływać na wzrost ryzyka raka piersi (43, 44, 45) natomiast zmniejszają o około 50% ryzyko raka jajnika (34). Tak więc ich stosowanie w późniejszym wieku wydaje się uzasadnione. Jak dotąd brak dobrze zweryfikowanych danych na temat efektów stosowania doustnej antykoncepcji w rodzinach z rakami piersi/jajnika niezwiązanymi z BRCA1. W związku z tym jednak, że istnieją prace o kilkakrotnym zwiększeniu ryzyka raka piersi spowodowanym pigułkami antykoncepcyjnymi u kobiet z rodzin z agregacją zachorowań na raka piersi (46), rozsądnym wydaje się unikanie doustnej antykoncepcji w rodzinach z cechami HBC-ss, HBOC czy też HOC.
Hormonalna terapia zastępcza
Profilaktyczne usunięcie narządu rodnego w wieku 35-40 lat jest postępowaniem z wyboru u nosicielek mutacji BRCA1/2i wiąże się z redukcją ryzyka rozwoju zarówno raka jajnika jak i raka piersi. Wykazano, że nosicielki mutacji po andexektomii, stosujące hormonalną terapię zastępczą opartą o estrogeny doświadczają podobnego efektu ochronnego jak pacjentki, które nie stosowały hormonalnej terapii zastępczej (47, 48). Wpływ hormonalnej terapii zastępczej u nosicielek mutacji BRCA1/2, które nie poddały się operacji profilaktycznej narządu rodnego nie jest wystarczająco udokumentowany. Stwierdzono 3-krotny – wzrost ryzyka rozwoju raka piersi u osób stosujących hormonalną terapię zastępczą i obciążonych rodzinnym wywiadem odnośnie raka piersi (49). W związku z powyższym decyzja o stosowaniu hormonalnej terapii zastępczej u takich nosicielek mutacji BRCA1/2, powinna być rozważana ze szczególną ostrożnością.
Karmienie piersią
Długotrwałe karmienie piersią zalecamy wszystkim pacjentkom z rodzin z HBC-ss, HBOC i HOC. Wykazano, że u nosicielek mutacji BRCA1karmienie przez okres, łącznie po wszystkich ciążach, długości 18 miesięcy redukuje ryzyko raka piersi o około 50% tj. z 50-80% do 25-40% (50, 51).
Wczesne urodzenie dziecka
Generalnie kobiety, które urodziły pierwsze dziecko przed 20 r.ż. mają o około połowę niższe od nieródek ryzyko zachorowania na raka piersi. Ta obserwacja prawdziwa w odniesieniu do nieselekcjonowanej grupy kobiet, nie została potwierdzona w grupie kobiet – nosicielek mutacji BRCA1czy BRCA2 (52). Biorąc jednak pod uwagę fakt, że nosicielki mutacji powinny poddać się operacji profilaktycznej narządu rodnego w wieku 35-40 lat, pacjentki nie powinny zwlekać z macierzyństwem.
Chemoprewencja
Tamoxifen
Dane z piśmiennictwa jednoznacznie wskazują, że tamoxifen zmniejsza o około 50% ryzyko raka piersi ER+. Działanie to zaobserwowano zarówno u zdrowych kobiet jak i u kobiet leczonych z powodu raka jednej piersi, kiedy to tamoxifen zmniejszał ryzyko raka obustronnego. Tamoxifen działa też profilaktycznie u nosicielek mutacji BRCA1obniżając ryzyko raka piersi o około 50% mimo tego, że zdecydowana większość tych guzów jest ER-. Dobroczynne działanie tamoxifenu wykazano zarówno u nosicielek mutacji w wieku przedmenopauzalnym, jak i pomenopauzalnym (53, 54). Wg aktualnych danych uzasadnione jest proponowanie pacjentkom z rodzin z definitywnym HBC-ss, HBOC oraz nosicielkom mutacji BRCA15-letniej chemoprewencji tamoxifenem po wykluczeniu wszelkich przeciwwskazań zwłaszcza związanych ze zwiększonym ryzykiem choroby zakrzepowej i przy zapewnieniu odpowiedniej kontroli, co do powstawania tych zaburzeń oraz zmian przerostowych śluzówki trzonu macicy.
Selen
Badania przeprowadzone w naszym Ośrodku wykazały, że u nosicielek mutacji BRCA1występuje zwiększona podatność na mutageny mierzona testem bleomycynowym. Podatność tą można normalizować za pomocą niektórych preparatów selenu (55). Chemoprewencja selenem została opisana jako działanie zmniejszające ryzyko różnych nowotworów u ludzi jak i u zwierząt doświadczalnych. Tak więc celowe wydaje się proponowanie nosicielkom mutacji BRCA1udziału w prowadzonym przez nas programie profilaktycznym.
Adnexektomia
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Lynch HT: Genetics and breast cancer. Van Nostrand – Reinhold, New York 1981.
2. Broca P: Traite de tumeurs. Paris, Asselin 1866.
3. Miki Y, et al.: A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266: 66-71.
4. Thompson D, Easton D:Breast Cancer Linkage Consortium. Variation in BRCA1 cancer risks by mutation position. Cancer Epidemiol Biomarkers Prev 2002; 11: 329-336.
5. Lichtenstein P, et al.: Environmental and heritable factors in the causation of cancer-analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000; 343: 78-85.
6. Swift M, et al.: Breast and other cancers in families with ataxia-telangiectasia. N Engl J Med 1987; 316: 1289-1294.
7. Antoniou AC, et al.: Breast and ovarian cancer risks to carriers of the BRCA1 5382insC and 185delAG and BRCA2 6174delT mutations: a combined analysis of 22 population based studies. J Med Genet 2005; 42: 602-603.
8. Gronwald J, et al.: Cancer risks in first degree relatives of BRCA1 mutation carriers: effects of mutation and proband disease status. J Med Genet 2006; 43: 424-428.
9. Risinger JI, et al.: Mutations of the E-cadherin gene in human gynecologic cancers. Nat Genet 1994; 7: 98-102.
10. Thorlacius S, et al.: Populationbased study of risk of breast cancer in carriers of BRCA2 mutation. Lancet 1998; 352: 1337-1339.
11. Rebbeck TR, et al.: The Prevention and Observation of Surgical End Points Study Group. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 2002; 346: 1616-1622.
12. Jakubowska A, et al.: Breast cancer risk reduction associated with the RAD51 polymorphism among carriers of the BRCA1 5382insC mutation in Poland. Cancer Epidemiol Biomarkers Prev 2003; 12: 457-459.
13. Jakubowska A, et al.: The RAD51 135 G>C polymorphism modifies breast cancer and ovarian cancer risk in Polish BRCA1 mutation carriers. Cancer Epidemiol Biomarkers Prev 2007; 16: 270-275.
14. Risch HA, et al.: Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 2001; 68: 700-710.
15. Marcus JN, et al.: Hereditary breast cancer: pathobiology, prognosis, and BRCA1 and BRCA2 gene linkage. Cancer 1996; 77: 697-709.
16. Martin AM, et al.: Germline mutations in BRCA1 and BRCA2 in breast-ovarian families from a breast cancer risk evaluation clinic. J Clin Oncol 2001; 19: 2247-2253.
17. Li FP, Fraumeni JF Jr: Soft-tissue sarcomas, breast cancer and other neoplasms. A familial syndrome? Ann Intern Med 1969; 71: 747-752.
18. Menkiszak J, et al.: Hereditary ovarian cancer: summary of 5 years of experience. Ginekol Pol 1998; 69: 283-287.
19. Loman N, et al.: Prognosis and clinical presentation of BRCA2-associated breast cancer. Eur J Cancer 2000; 36: 1365-1373.
20. Lubinski J, et al.: BRCA1-positive breast cancers in young women from Poland. Breast Cancer Res Treat 2006; 99: 71-76.
21. Lakhani SR: The pathology of familial breast cancer: Morphological aspects. Breast Cancer Res 1999; 1: 31-35.
22. Górski B, et al.: Founder mutations in the BRCA1 gene in Polish families with breast-ovarian cancer. Am J Hum Genet 2000; 66: 1963-1968.
23. Loman N, et al.: Steroid receptors in hereditary breast carcinomas associated with BRCA1 or BRCA2 mutations or unknown susceptibility genes. Cancer 1998; 83: 310-319.
24. Wooster R, et al.: Localisation of a breast cancer susceptibility gene BRCA2, to chromosome 13q12-13. Science 1994; 265: 2088-2090.
25. Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 in a population based series of breast cancer cases. Br J Cancer 2000; 83: 1301-1308.
26. Ford D, et al.: Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62: 676-689.
27. Hopper JL, et al.: Australian Breast Cancer Family Study. Population-based estimate of the average agespecific cumulative risk of breast cancer for a defined set of protein-truncating mutations in BRCA1 and BRCA2. Cancer Epidemiol Biomarkers Prev 1999; 8: 741-747.
28. Warner E, et al.: Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst 1999; 91: 1241-1247.
29. Jakubowska A, et al.: BRCA2 gene mutation in familie with aggregations of breast and stomach cancers. Br J Cancer 2002; 87: 888-891.
30. Jakubowska A, et al.: A high frequency of BRCA2 gene mutations in Polish families with ovarian and stomach cancer. Eur J Hum Genet 2003; 11: 955-958.
31. Kwiatkowska E, et al.: BRCA2 germline mutations in male breast cancer patients in the Polish population. Hum Mutat 2001; 17: 73.
32. Boyd J, et al.: Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA 2000; 283: 2260-2265.
33. Malkin D, et al.: Germline p53 mutations in a familiar syndrome of breast cancer, sarcomas and other neoplasms. Science 1990; 250: 1233-1238.
34. Nelen MR, et al.: Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet 1996; 13: 114-116.
35. Hanssen AM, Fryns JP: Cowden syndrome. J Med Genet 1995; 32: 117-119.
36. Risinger JI, et al.: Molecular genetic evidence of the occurrence of breast cancer as on integral tumor in patients with the hereditary nonpolyposis colorectal carcinoma syndrome. Cancer 1996; 77: 1836-1843.
37. Spigelman AD, Murday V, Phillips RK: Cancer and Peutz-Jeghers syndrome. Gut 1989; 30: 1588-1590.
38. Cohen MM Jr: A comprehensive and critical assessment of overgrowth and overgrowth syndromes. Adv Hum Genet 1989; 18: 181-303, 373-376.
39. Shiloh Y: Ataxia telangiectasia: closer to unraveling the mystery. Eur J Hum Genet 1995; 3: 116-138.
40. Lynch HT, Kaplan AR, Lynch JF: Klinefelter syndrome and cancer. A family study. JAMA 1974; 229: 809-811.
41. Wooster R, et al.: A germline mutation in the androgen receptor in two brothers with breast cancer and Reifenstein syndrome. Nat Genet 1992; 2: 132-134.
42. Lindblom A, et al.: Predisposition for breast cancer in carriers of constitutional translocation 11q;22q. Am J Hum Genet 1994; 54: 871-876.
43. Narod SA, et al.: Oral contraceptives and the risk of hereditary ovarian cancer. Hereditary Ovarian Cancer Clinical Study Group. N Engl J Med 1998; 339: 424-428.
44. McLaughlin JR, et al.: Hereditary Ovarian Cancer Clinical Study Group. Reproductive risk factors for ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet Oncol 2007; 8: 26-34.
45. Narod SA, et a.: Oral contraceptives and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 2002; 94: 1773-1779.
46. Grabrick DM, et al.: Risk of breast cancer with oral contraceptive use in women with a family history of breast cancer. JAMA 2000; 284: 1791-1798.
47. Armstrong K, et al.: Hormon replacement therapy and life expectancy after prophylactic oophorectomy in women with BRCA1/2 mutations: a decision analysis. J Clin Oncol 2004; 22: 1045-1054.
48. Rebbeck TR, et al.: Effect of Short-Term Hormone Replacement Therapy on Breast Cancer Risk Reduction After Bilateral Prophylactic Oophorectomy in BRCA1 and BRCA2 Mutation Carriers: The PROSE Study Group. J Clin Oncol 2005; 23: 7804-7810.
49. Buchet-Poyau K, et al.: Search for the second Peutz-Jeghers syndrome locus: exclusion of the STK13, PRKCG, KLK10, and PSCD2 genes on chromosome 19 and the STK11IP gene on chromosome 2. Cytogenet Genome Res 2002; 97: 171-178.
50. Gronwald J, et al.: Influence of selected lifestyle factors on breast and ovarian cancer risk in BRCA1 mutation carriers from Poland. Breast Cancer Res Treat 2006; 95: 105-109.
51. Narod SA: Hormonal prevention of hereditary breast cancer. Ann N Y Acad Sci 2001; 952: 36-43.
52. Kotsopoulos J, et al.: Age at first birth and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat 2007; 105: 221-228.
53. Gronwald J, et al.: Hereditary Breast Cancer Clinical Study Group. Tamoxifen and contralateral breast cancer in BRCA1 and BRCA2 carriers: an update. Int J Cancer 2006; 118: 2281-2284.
54. Narod SA, et al.: Hereditary Breast Cancer Clinical Study Group. Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case-control study. Hereditary Breast Cancer Clinical Study Group. Lancet 2000; 356: 1876-1881.
55. Kowalska E, et al.: Increased rates of chromosome breakage in BRCA1 carriers are normalized by oral selenium supplementation. Cancer Epidemiol Biomarkers Prev 2005; 14: 1302-1306.
56. Menkiszak J, et al.: Attitudes toward preventive oophorectomy among BRCA1 mutation carriers in Poland. Eur J Gynaecol Oncol 2004; 25: 93-95.
57. Zeigler LD, Kroll SS: Primary breast cancer after prophylactic mastectomy. Am J Clin Oncol 1991; 14: 451-454.
58. Temple WJ, et al.: Technical considerations for prophylactic mastectomy in patients at high risk for breast cancer. Am J Surg 1991; 161: 413-415.
59. Warner E, et al.: Comparison of breast magnetic resonance imaging, mammography, and ultrasound for surveillance of women at high risk for hereditary breast cancer. J Clin Oncol 2001; 19: 3524-3531.
60. Narod SA, et al.: Oral contraceptives and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 2002; 94: 1773-1779.
61. Byrski T, et al.: Response to neo-adjuvant chemotherapy in women with BRCA1-positive breast cancers. Breast Cancer Res Treat 2008; 108: 289-296.
62. Lubinski J, et al.: Genetic contribution to all cancers: the first demonstration using the model of breast cancers from Poland stratified by age at diagnosis and tumour pathology. Breast Cancer Res Treat 2008 Apr 15.
63. Cybulski C, et al.: CHEK2-positive breast cancers in young Polish women. Clin Cancer Res 2006; 12: 4832-4835.
64. Cybulski C, et al.: A deletion in CHEK2 of 5,395 bp predisposes to breast cancer in Poland. Breast Cancer Res Treat 2007; 102: 119-122.
65. Huzarski T, et al.: Pathology of breast cancer in women with constitutional CHEK2 mutations. Breast Cancer Res Treat 2005; 90: 187-189.
66. Steffen J, et al.: Germline mutations 657del5 of the NBS1 gene contribute significantly to the incidence of breast cancer in Central Poland. Int J Cancer 2006; 119: 472-475.
67. Górski B, et al.: Germline 657del5 mutation in the NBS1 gene in breast cancer patients. Int J Cancer 2003; 106: 379-381.
68. Huzarski T, et al.: The 3020insC allele of NOD2 predisposes to earlyonset breast cancer. Breast Cancer Res Treat 2005; 89: 91-93.
69. Górski B, Narod SA, Lubinski J: A common missense variant in BRCA2 predisposes to early onset breast cancer. Breast Cancer Res 2005; 7: R1023-R1027.
70. Menkiszak J, et al.: Clinical features of familial ovarian cancer lacking mutations in BRCA1 or BRCA2. Eur J Gynaecol Oncol 2004; 25: 99-100.
71. Hart WR: Mucinous tumors of the ovary: A review. Int J Gynecol Pathol 2005; 24: 4-25.
72. Shih IeM, Kurman RJ: Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol 2004; 164: 1511-1518.
73. Szymańska A: Identyfikacja genów związanych z predyspozycji do gruczolako-torbielaków oeluzowych jajnika. Doctor´s thesis. Pomorska Akademia Medyczna, Szczecin 2006.
74. Szymańska-Pasternak J: Identyfikacja genów związanych z predyspozycją do gruczolako-torbielaków surowiczych jajnika. Doctor´s thesis. Pomorska Akademia Medyczna, Szczecin 2007.
