© Borgis - Postępy Nauk Medycznych 7/2008, s. 472-481
*Barbara Jarząb1, Jan Włoch2, Zbigniew Wygoda1
Genetyka kliniczna raka rdzeniastego tarczycy
Clinical genetic of medullary thyroid carcinoma
1Zakład Medycyny Nuklearnej i Endokrynologii Onkologicznej, Centrum Onkologii-Instytut im. Marii Skłodowskiej-Curie, Gliwice
Kierownik Zakładu: prof. dr hab. med. Barbara Jarząb
2Klinika Chirurgii Onkologicznej i Rekonstrukcyjnej, Centrum Onkologii-Instytut im. Marii Skłodowskiej-Curie, Gliwice
Kierownik Kliniki: dr lek. med. Stanisław Półtorak
Streszczenie
Rak rdzeniasty tarczycy (RRT) jest neuroendokrynnym nowotworem złośliwym, wywodzącym się z okołopęcherzykowych komórek C. Komórki te pochodzą z grzebienia nerwowego, w czasie rozwoju płodowego migrują z V kieszonki skrzelowej do tarczycy, gdzie produkują kalcytonine. Kalcytonina jest hormonem peptydowym, ułatwiającym przejście wapnia z krwi do kości.
Rak rdzeniasty tarczycy występuje w postaci sporadycznej oraz dziedzicznej, której wystąpienie związane jest z obecnością mutacji protoonkogenu RET. Dziedzicznemu RRT mogą nie towarzyszyć żadne inne objawy i mówi się wówczas o rodzinnym raku rdzeniastym tarczycy (ang. familial medullary thyroid carcinoma, FMTC). Częściej jednak dziedziczny RRT jest objawem zespołu gruczolakowatości wewnątrzwydzielniczej typu 2 (ang. multiple endocrine neoplasia type 2, MEN 2).
Zespół gruczolakowatości wewnątrzwydzielniczej typu 2A (MEN 2A), zwany również zespołem Sipple´a, charakteryzuje się skojarzeniem RRT z guzami chromochłonnymi nadnerczy (u około 50% chorych) i gruczolakami lub hiperplazją przytarczyc (u około 15-25% chorych). Rozpoznanie zespołu MEN 2B jest daleko bardziej jednoznaczne, tak ze względu na charakterystyczny obraz kliniczny jak i charakterystyczne mutacje. W tym zespole RRT rozwija się najszybciej, jeszcze u małych dzieci. Guzy chromochłonne nadnerczy występują później i ujawniają się u około połowy chorych, natomiast nadczynność przytarczyc nie występuje.
W pracy przedstawiono aktualny stan wiedzy na temat molekularnego podłoża dziedzicznej postaci RRT oraz zalezność między lokalizacją mutacji punktowej RET i obrazem klinicznym choroby. Omówiono również postępowanie diagnostyczne i lecznicze w dziedzicznej postaci RRT oraz postępowanie w razie wykrycia nosicielstwa mutacji protoonkogenu RET. Jednocześnie w podsumowaniu podano krótkie wskazówki dotyczące postępowania w przypadku wykrycia dziedzicznej postaci RRT.
Summary
Medullary thyroid carcinoma (MTC) is neuroendocrine malignant neoplasm, arising from the parafollicular thyroid cells. These cells arising from the neural crest, migrating from the fifth branchial cleft into thyroid gland during the embryogenesis, where the calcitonin hormone is producing by them. Calcitonin is the peptide hormone, facilitating calcium transition from the blood to the bones.
Medullary thyroid carcinoma occurs in the sporadic and hereditary form, which presence is connected with protooncogene RET mutations. Hereditary form of MTC can be divided into familial medullary thyroid carcinoma (FMTC) without any endocrinopathies and, more frequently, as a part of multiple endocrine neoplasia type 2 (MEN 2).
Multiple endocrine neoplasia type 2A (MEN 2A), named as Sipple synrome also, can be characterized as presence of MTC and pheochromocytoma (in about 50% of patients) and parathyroid adenomas or hyperplasia (15-25% of patients). Recognition of syndroma MEN2B is more unequivocal because of characteristic clinical status and characteristic mutations. In this syndrome, MTC develops the most quickly, even in young children. Pheochromocytomas occur later and in the half of patients, parathyroid adenomas are absent.
In this paper actual state of knowledge about the molecular basis of hereditary form of MTC and dependence between localization of RET mutations and clinical disease status are presented. Diagnostic and therapeutic procedures in hereditary form of MTC and the way of proceeding in the case of RET mutation presence are discussed Short guidelines about management in the case of hereditary form of MTC are presented also.

Rak rdzeniasty tarczycy jest neuroendokrynnym nowotworem złośliwym, wywodzącym się z okołopęcherzykowych komórek C. W piśmiennictwie jego odkrycie wiąże się z nazwiskiem Hazarda (1). Należy jednak podkreślić, że pierwsze doniesienie opisujące ten typ raka zostało opublikowane w piśmiennictwie polskim, w „Nowotworach” przez prof. Laskowskiego, który nazwał ten typ nowotworu „ca hyalinicum”.
Komórki C pochodzą z grzebienia nerwowego, w czasie rozwoju płodowego migrują z V kieszonki skrzelowej do tarczycy, gdzie produkują kalcytoninę. Kalcytonina jest hormonem peptydowym, ułatwiającym przejście wapnia z krwi do kości.
Rak rdzeniasty tarczycy (RRT) zlokalizowany jest najczęściej w środkowo-górnej części bocznych płatów tarczycy, gdzie nagromadzenie komórek okołopęcherzykowych jest największe. Komórki raka zwykle ułożone są w gniazda, oddzielone od siebie cienkimi warstwami włóknisto-naczyniowego zrębu, rzadziej tworzą beleczki, wysepki lub przyjmują utkanie lite. W otaczającym miąższu tarczycy mogą być widoczne cechy hiperplazji komórek C. Cechą charakterystyczną, ale nie zawsze stwierdzaną, jest obecność amyloidu, dlatego rozpoznanie histopatologiczne raka rdzeniastego wymaga, obok klasycznego badania mikroskopowego, badania immunohistochemicznego, przede wszystkim zastosowania przeciwciał przeciw kalcytoninie. W ponad 90% przypadków obecność RRT wiąże się ze znacznym wzrostem stężenia kalcytoniny (Ct) w surowicy krwi. Dlatego oznaczenie kalcytoniny we krwi u chorych z podejrzeniem RRT ułatwia jego rozpoznanie.
Rak rdzeniasty tarczycy szerzy się zarówno drogą chłonną, jak i krwionośną. Przerzuty do węzłów chłonnych stwierdza się w czasie rozpoznania w 50-75% przypadków, często obustronnie, z naciekami pozatorebkowymi. Rozsiew węzłowy dotyczy na ogół najpierw węzłów przedtchawiczych, a dopiero następnie bocznych szyi i nie zawsze można go uwidocznić w badaniu ultrasonograficznym. Stopień zajęcia węzłów chłonnych zwykle koreluje z wielkością ogniska pierwotnego. Drogą krwionośną powstają przerzuty do wątroby, płuc i kości.
W przypadku zaawansowanego miejscowo raka rdzeniastego tarczycy naciek nowotworowy może szerzyć się przez ciągłość poza torebkę narządu, obejmując pęczki naczyniowo-nerwowe, mięśnie szyi, a także tchawicę i przełyk.
Pierwszym objawem RRT u większości chorych jest guzek tarczycy, stopniowo powiększający się, o różnej dynamice wzrostu, najczęściej wolnej, z reguły bezbolesny. U kilku – kilkunastu procent chorych występuje biegunka, która czasem może być pierwszym objawem RRT i wiąże się z zaawansowaną postacią nowotworu, a spowodowana jest wydzielaniem przez guz czynnych biologicznie peptydów i amin. Przy znacznym zaawansowaniu miejscowym choroby pojawia się duszność, uczucie przeszkody przy połykaniu lub wręcz zaburzenia połykania. Kaszel, powiększenie wątroby, bolesność samoistna i uciskowa kośćca, szybka utrata wagi ciała mogą towarzyszyć rozsianej postaci nowotworu. RRT należy do tych nowotworów, w których udział predyspozycji dziedzicznej jest stosunkowo wysoki i wynosi 20-25% wszystkich przypadków, a w populacjach objętych intensywnymi badaniami przesiewowymi wśród członków rodzin nawet ponad 30% przypadków (2, 3).
Dziedziczny rak rdzeniasty tarczycy
Dziedzicznemu RRT mogą nie towarzyszyć żadne inne objawy i mówimy wówczas o rodzinnym raku rdzeniastym tarczycy (ang. familial medullary thyroid carcinoma, FMTC). Częściej jednak dziedziczny RRT jest objawem zespołu gruczolakowatości wewnątrzwydzielniczej typu 2 (ang. multiple endocrine neoplasia type 2, MEN 2) (tab. 1).
Tabela 1. Dziedziczny rak rdzeniasty tarczycy: postaci kliniczne.
| Objaw | FMTC | MEN 2A | MEN 2B |
| rak rdzeniasty tarczycy | > 95% | > 95% | > 95% |
| guz chromochłonny | - | ~ 50% | ~ 50% |
| nadczynność przytarczyc | - | 15-60% | - |
| typowy wygląd twarzy, nerwiaki błon śluzowych, przerost zwojów przywspółczulnych, błony śluzowej jelita grubego | - | - | 100% |
Zespół gruczolakowatości wewnątrzwydzielniczej typu 2A (MEN 2A), zwany również zespołem Sipple´a, charakteryzuje się skojarzeniem RRT z guzami chromochłonnymi nadnerczy (u około 50% chorych) i gruczolakami lub hiperplazją przytarczyc (u około 15- 25% chorych). RRT jest zwykle pierwszym objawem zespołu i ujawnia się w pierwszych dwu dekadach życia. Guzy chromochłonne nadnerczy na ogół ujawniają się później i rzadko są pierwszym objawem choroby. Najpóźniej dochodzi do ujawnienia nadczynności przytarczyc, dlatego też ocena jej występowania różni się w zależności od wieku chorych w badanej populacji.
W nietypowych postaciach zespołu MEN 2A towarzyszą mu także liszaj amyloidowy (ang. cutaneous lichen amyloidosis, CLA) lub choroba Hirschsprunga, są to jednak zespoły stosunkowo rzadkie (2).
Guzy chromochłonne nadnerczy charakteryzują się napadowym nadciśnieniem tętniczym, przebiegającym z tachykardią, może im towarzyszyć zblednięcie i nadmierne pocenie się. Nierozpoznany/nieleczony guz chromochłonny może być przyczyną nagłej śmierci i stanowi większe nawet zagrożenie dla życia chorego niż RRT, który w zespole MEN 2A może przebiegać mało agresywnie.
Nadczynność przytarczyc prowadzi do wzrostu poziomu wapnia w surowicy krwi, wywołanego nadmiarem parathormonu. Parathormon nasila resorpcję kostną, stąd wczesnym objawem jest osteoporoza; objawy resorpcji podokostnowej, czy guzy brunatne pojawiają się znacznie później. Do cech zaawansowanej nadczynności przytarczyc należą objawy kamicy nerkowej, może pojawić się choroba wrzodowa żołądka i zapalenie trzustki. Nieleczona nadczynność przytarczyc może doprowadzić do przełomu hiperkalcemicznego.
Ponieważ RRT ujawnia się najwcześniej, odróżnienie rodzinnego RRT od klasycznego zespołu MEN 2A wymaga dłuższej obserwacji, gdyż guzy chromochłonne mogą się ujawnić po latach i nigdy nie wystąpią u wszystkich członków rodziny, u których rozwinął się RRT. W piśmiennictwie przyjmuje się na ogół, że rozpoznanie prawdziwego raka rodzinnego jest pewne dopiero, kiedy w rodzinie są już co najmniej 4 przypadki RRT, którym nie towarzyszy ani guz chromochłonny tarczycy ani nadczynność przytarczyc. Jeżeli liczba chorych na RRT jest mniejsza od 4, mówimy o postaci niesklasyfikowanej, gdyż nawet test DNA nie pozwala na jednoznaczne różnicowanie w tym zakresie (patrz niżej).
Rozpoznanie zespołu MEN 2B jest daleko bardziej jednoznaczne, tak ze względu na charakterystyczny obraz kliniczny jak i charakterystyczne mutacje. W tym zespole RRT rozwija się najszybciej, jeszcze u małych dzieci. Guzy chromochłonne nadnerczy występują później i ujawniają się u około połowy chorych, natomiast nadczynność przytarczyc nie występuje. Cechy fenotypowe zespołu MEN 2B pozwalają doświadczonemu klinicyście na rozpoznanie już przy pierwszym kontakcie z chorym. Jego wygląd jest niezwykle charakterystyczny, z podłużną wąską twarzą, dużą żuchwą i bardzo wydatnymi ustami. Stwierdzenie licznych drobnych nerwiaków na brzegach języka i śluzówce jamy ustnej jest bardzo swoistą cechą w badaniu fizykalnym. U części chorych zaznaczają się też marfanoidalne cechy budowy ciała. Dziedziczny charakter części przypadków RRT był znany już od lat sześćdziesiątych XX wieku. Dla wczesnego wykrywania raka wśród członków rodziny chorego stosowano oznaczenie kalcytoniny po podaniu pentagastryny (4). Badania takie przeprowadzano co roku wśród wszystkich członków rodziny do czasu osiągnięcia przez nich czterdziestego roku życia. Dla uniknięcia wyników fałszywie dodatnich (pentagastryna może stymulować wzrost wydzielania kalcytoniny także u zdrowych osób, szczególnie u młodych mężczyzn) jako patognomoniczny dla dziedzicznego RRT traktowano wzrost kalcytoniny powyżej 100 pg/ml. Oznaczenie stężenia kalcytoniny umożliwiło dobrą charakterystykę predyspozycji genetycznej wśród członków rodzin, ułatwiło więc badanie sprzężenia między występowaniem RRT i markerami genetycznymi.
Protoonkogen RET i rak rdzeniasty tarczycy
W 1987 roku zlokalizowano gen odpowiedzialny za dziedziczne postaci RRT w centromerowym regionie chromosomu 10. W 1993 zidentyfikowano go jako protoonkogen RET oraz opisano mutacje odpowiedzialne za zespół MEN 2A, FMTC i zespół MEN 2B (5, 6, 7).
Protoonkogen RET koduje receptorową kinazę tyrozynową. W części zewnątrzkomórkowej tego białka błonowego znajduje się region podobny do kadheryny oraz zlokalizowany blisko błony komórkowej region bogaty w cysteinę (ryc. 1).
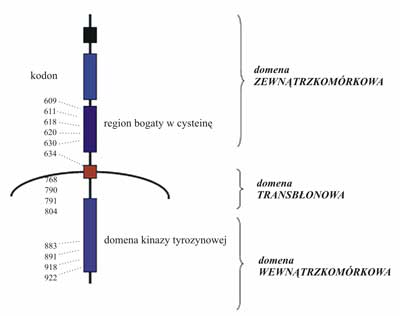
Ryc. 1. Schemat budowy receptora kinazy tyrozynowej RET wraz z lokalizacją kodonów podlegających mutacjom aktywującym.
Krótka część przezbłonowa utrzymuje białko w błonie komórkowej, a w części wewnątrzcytoplazmatycznej znajduje się domena, a właściwie dwie blisko położone domeny o aktywności kinazy tyrozynowej. Budowa białka ściśle nawiązuje do budowy innych receptorów dla czynników wzrostowych (np. EGF), które są de facto receptorowymi kinazami tyrozynowymi.
Ligandem odpowiedzialnym za przekazywanie sygnału wzrostowego poprzez białko RET jest niewielki neuropeptyd, glejopochodny czynnik neurotropowy (ang. glial cell-derived neurotrophic factor, GDNF). Peptyd ten nie łączy się bezpośrednio z białkiem RET, a z innym białkiem błonowym, zwanym receptorem α dla GDNF (obecnie GFRα-1), pełniącym funkcję koreceptora dla RET (ryc. 2). Następstwem aktywacji receptora jest jego autofosforylacja, uruchamiająca kaskadę kinaz MAP i transkrypcję genów uczestniczących w proliferacji komórkowej.
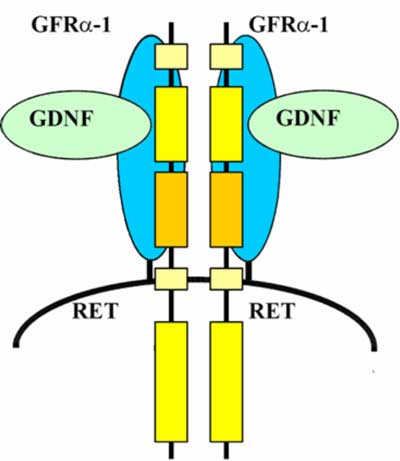
Ryc. 2. Fizjologiczna aktywacja kinazy tyrozynowej RET.
Mutacje protoonkogenu RET, prowadzące do rozwoju RRT, mają charakter mutacji aktywujących funkcję produktu białkowego (2). Protoonkogen RET składa się z 21 eksonów. Mutacje występują jednak zaledwie w kilku z nich i w przeważającej większości mają charakter mutacji punktowych (na rycinie 1 ukazano ich lokalizację w odniesieniu do kodowanego białka). Najczęściej dotyczą one kodonów kodujących cysteiny w części zewnątrzbłonowej receptora, blisko błony komórkowej. W przeważającej liczbie (w 75-80% wszystkich przypadków dziedzicznego RRT) ulega mutacji kodon 634, zlokalizowany w eksonie 11, wchodzącym już w skład części przezbłonowej (tab. 2). Większość mutacji w tym kodonie (ponad 90%) stanowią mutacje, których następstwem jest zamiana cysteiny na argininę, tyrozynę lub tryptofan (8, 9).
Tabela 2. Lokalizacja mutacji protoonkogenu RET powodujących dziedzicznego raka rdzeniastego tarczycy (10, 11).
| Kodon/Ekson | Zespół | Częstość występowania (13) | Częstość występowania (%) (39) |
| 609/10 | MEN 2A/FMTC
MEN 2A/ch.
Hirschsprunga | 0-1 | 0% |
| 611/10 | MEN 2A/FMTC | 2-3 | 2,5% |
| 618/10 | FMTC/MEN 2A
MEN 2A/ch.
Hirschsprunga | 3-5 | 12% |
| 620/10 | FMTC/MEN 2A
MEN 2A/ch.
Hirschsprunga | 6-8 | 3% |
| 630/11 | MEN 2A/FMTC | 0-1 | 0% |
| 634/11 | MEN 2A
MEN 2A/CLA | 75-85 | 42% |
| 635/11 | MEN 2A | rzadko | nie badano |
| 637/11 | MEN 2A | rzadko | nie badano |
| 768/13 | FMTC | 0-1 | 1% |
| 790/13 | FMTC/MEN 2A | 0-1 | 2,5% |
| 791/13 | FMTC | 0-1 | 16% |
| 804/13 | MEN 2A/FMTC | 0-1 | 8% |
| 883/15 | MEN 2B | rzadko | rzadko |
| 891/15 | FMTC | rzadko | nie badano |
| 918/16 | MEN 2B | 3-5 | 12% |
| 922/16 | MEN 2B | rzadko | nie badano |
Klasyczny zespół MEN 2A jest najbardziej prawdopodobny, jeżeli mamy do czynienia z mutacją w kodonie 634, podczas gdy inne mutacje wiążą się ze znacznie mniejszym prawdopodobieństwem rozwoju guza chromochłonnnego – najczęściej w jej wyniku rozwija się zespół rodzinnego RRT bez innych endokrynopatii (tab. 2).
Mutacje w kodonie 918 (ekson 16) dotyczą domeny kinazy tyrozynowej. Ponieważ fosforylacji ulegają nieco inne białka komórkowe, fenotyp zespołu MEN 2B różni się od fenotypu zespołu MEN 2A. Nadmierna aktywacja RET uwidacznia się także w nerwach obwodowych (nerwiaki języka i błony śluzowej jamy ustnej i warg, hyperganglionoza jelita grubego), rak rdzeniasty ujawnia się wcześniej i ma bardziej agresywny przebieg, nie dochodzi natomiast do hiperplazji przytarczyc (12, 13).
Mutacje w kodonach 768, 790, 791, 804 i 891 dotyczą także części wewnątrzkomórkowej białka RET (14, 15, 16, 17), występują rzadko, a ich potencjał transformujący jest niewielki – poza mutacją w kodonie 790, którą wykazano tak w FMTC jak i MEN 2A (14), prowadzą głównie do rozwoju rodzinnego RRT, który może ujawnić się stosunkowo późno – często dopiero w czwartej – piątej dekadzie życia. W odniesieniu do mutacji w kodonie 791 podnoszone są przypuszczenia, że jej penetracja może być niepełna. Pozostałe mutacje genu RET charakteryzują się prawie pełną penetracją – stwierdzenie mutacji germinalnej jest więc równoznaczne z ponad 90% pewnością rozwoju RRT. W rodzinach z mutacją RET 791 istnieje duża zmienność ryzyka i co najmniej w niektórych z nich stosunkowo wcześnie ujawnia się pełnoobjawowy RRT.
Zależność genotyp-fenotyp w postaci dziedzicznej raka rdzeniastego tarczycy
W dziedzicznym RRT da się wyraźnie wyodrębnić zależność między lokalizacją mutacji punktowej RET i obrazem klinicznym choroby.
Z genetycznego punktu widzenia, zespół MEN 2A i rodzinny RRT są do siebie zbliżone i dzisiaj raczej traktuje się te zespoły łącznie – rodzinny RRT jest jedną z postaci zespołu MEN 2A. Zespół MEN 2B wydziela się osobno, tak ze względu na typowe mutacje jak i typowy fenotyp. Prawdopodobieństwo wystąpienia typowego zespołu MEN 2A silnie zależy od lokalizacji mutacji – jest bardzo wysokie, jeżeli mutacja dotyczy kodonu 634 (przy czym pełen zespół, z nadczynnością przytarczyc, występuje szczególnie często przy podstawieniu cysteiny argininą), niższe w eksonie 10, a bardzo niskie przy mutacji w eksonie 13 i 15 (tab. 2) (18).
Testy DNA
Ponieważ rak dziedziczny stanowi znaczącą część wszystkich przypadków RRT i może klinicznie nie odróżniać się od raka nie-dziedzicznego, pełne badanie molekularne w kierunku obecności mutacji germinalnych musi być przeprowadzone u wszystkich chorych, u których postawiono takie rozpoznanie. Nawet, jeżeli u chorego nie występują inne cechy zespołu MEN 2 i wywiad rodzinny jest ujemny, ryzyko wykrycia mutacji germinalnej wynosi w naszej populacji około 10% (3). Na rycinie 3 przedstawiono algorytm poszukiwania mutacji w protoonkogenie RET (19, 20). Kolejność badania kodonów protoonkogenu RET jest podyktowana częstością mutacji, zaczyna się więc od badania w kierunku mutacji w kodonie 634. To badanie może być wykonane techniką PCR/RFLP. Niemniej, ujemny wynik tego badania nie upoważnia do rezygnacji z badania w kierunku pozostałych znanych mutacji, gdyż blisko połowa nowo wykrytych przypadków raka dziedzicznego dotyczy mutacji w eksonach 13-15. Z tego samego powodu nie należy rezygnować z badania DNA przy rozpoznaniu RRT u osób starszych – jak już wspomniano, niektóre mutacje charakteryzują się wyraźnie późniejszym ujawnieniem RRT.
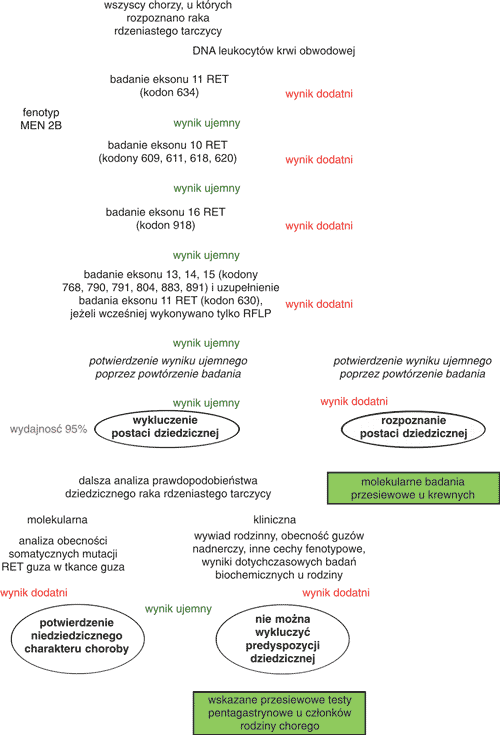
Ryc. 3. Algorytm diagnostyki DNA u chorych na raka rdzeniastego tarczycy.
Badanie mutacji w eksonach 10 i 13-15 wymaga sekwencjonowania. Badanie w kierunku mutacji w kodonie 918 wykonuje się na ogół na podstawie informacji o fenotypie, chociaż w części przypadków cechy fenotypowe zespołu MEN 2B mogą być słabo zaznaczone i dopiero badanie molekularne pozwala na postawienia prawidłowego rozpoznania. Należy podkreślić, że niewielki rozmiar genu i ograniczona liczba charakterystycznych mutacji są czynnikami ułatwiającymi badanie DNA. W naszym kraju badanie to przeprowadza kilka ośrodków1. Zidentyfikowanie mutacji germinalnej u chorego na RRT niesie istotne korzyści dla chorego i jego rodziny. U samego chorego pozwala określić ryzyko wystąpienia guza chromochłonnego nadnercza i nadczynności przytarczyc, determinuje więc częstość badań przesiewowych w tym kierunku. Jednocześnie, wykazanie predyspozycji dziedzicznej u chorego stanowi bezwzględne wskazanie do wdrożenia badań DNA u członków jego rodziny. Ryzyko wykrycia mutacji wśród krewnych pierwszego stopnia wynosi 50%. Nasze badania wskazują, że wykrywa się wówczas średnio co najmniej 1 nosiciela mutacji na każdy nowo wykryty przypadek dziedzicznego RRT (3, 21). Prawdopodobieństwo wczesnego wykrycia raka u nosiciela mutacji zmienia się w zależności od typu mutacji i wieku członków rodziny – u części badanych mamy już do czynienia z wykrywalnym klinicznie rakiem tarczycy, u innych stwierdzamy wzrost stężenia kalcytoniny albo w badaniu podstawowym albo po stymulacji pentagastryną, ale jeszcze bez widocznego w USG tarczycy guzka. Przy wczesnym wdrożeniu badań możliwe jest wykrycie nosicielstwa w stadium w pełni bezobjawowym.
Równie istotny jest negatywny wynik badania w kierunku mutacji germinalnej. Pozwala on wyłączyć danego członka rodziny z dalszych badań kontrolnych, jeżeli nie stwierdza się mutacji charakterystycznej dla danej rodziny. Natomiast negatywny wynik badania DNA, wykonywanego po rozpoznaniu raka rdzeniastego dla wykrycia postaci dziedzicznej, ma wartość predykcyjną około 90% (2). Istnieją bowiem rodziny (szczególnie dotyczy to rodzinnego RRT), u których pomimo kilku przypadków raka rdzeniastego nie udało się znaleźć mutacji germinalnej. Jeżeli więc wywiad rodzinny lub osobniczy jest dodatni, a test DNA jest ujemny, jedynym wyjściem jest kontynuacja corocznych testów pentagastrynowych u całej rodziny.
Badania biochemiczne stosowane w rozpoznaniu i monitorowaniu raka rdzeniastego tarczycy i zespołu MEN 2
Komórki RRT wydzielają na ogół duże ilości kalcytoniny. Jej oznaczanie daje możliwość przedoperacyjnej detekcji raka, a ponadto jest dobrym narzędziem oceny skuteczności zastosowanego leczenia i monitorowania dalszego przebiegu choroby (22, 23, 24). Obecnie rutynowe wykonywanie testów pentagastrynowych u członków rodziny chorego nie jest potrzebne, skoro postać dziedziczną można zidentyfikować badaniem DNA. Oznaczanie kalcytoniny powinno odbywać się w wyspecjalizowanej pracowni, dobre testy mają zakres normy do dziesięciu-kilkunastu pg/ml. Oznaczanie stężenia kalcytoniny jest niezbędnym elementem oceny skuteczności zastosowanego leczenia w raku rdzeniastym tarczycy. Normalizacja podwyższonego przedoperacyjnego stężenia hormonu do wartości prawidłowych po przebytej operacji potwierdza jej radykalność. Utrzymywanie się wartości nieprawidłowych, pomimo makroskopowej i mikroskopowej radykalności zabiegu, przemawia za obecnością mikroognisk raka w węzłach chłonnych.
Niskie stężenie hormonu (poniżej 10-12 pg/ml) w badaniach kontrolnych, wykonywanych co 3 miesiące, przemawia za całkowitą regresją guza. Konieczne jest jednak wykonanie raz do roku próby prowokacyjnej. Stosuje się w tym celu dożylne podanie wapnia, pentagastryny czy doustnie omeprazolu, przy czym próba pentagastrynowa jest stosowana najczęściej. Oznaczenie stężenia kalcytoniny przeprowadza się w próbkach pobranych w 2, 5 i 10 minut po podaniu pentagastryny (0,5 μg/kg masy ciała). Wzrost stężenia kalcytoniny powyżej 30 pg/ml świadczy o obecności komórek nowotworowych.
Wysokie stężenie kalcytoniny jest wskazaniem do badań obrazowych dla lokalizacji ogniska nowotworowego. Jeżeli ich wynik jest ujemny, można rozważyć cewnikowanie żył szyjnych i wątrobowych. Wzrost stężenia kalcytoniny w określonej próbce po podaniu pentagastryny pozwala zlokalizować wznowę lub przerzut w dorzeczu żyły, z której pobrano próbkę.
Stosunkowo rzadko obserwuje się prawidłowe stężenie kalcytoniny u chorych z jawnym klinicznie rakiem rdzeniastym. U części chorych może on wynikać z niskiego stopnia zróżnicowania nowotworu i utraty czynności hormonalnej. Należy też pamiętać, że wysokie stężenia antygenu (w tym przypadku kalcytoniny) może hamować jego wiązanie z przeciwciałem, używanym w teście radioimmunologicznym, przez co uzyskuje się wynik fałszywie ujemny (tzw. „hook effect”). Aby wykryć to zjawisko, wystarczy oznaczyć surowicę po rozcieńczeniu. Rozcieńczenie surowicy jest zresztą konieczne bardzo często, gdyż stężenia kalcytoniny obserwowane u chorych na raka rdzeniastego tarczycy mogą 10-1000 x przekraczać zakres stężeń oznaczalnych w dostępnych testach immunometrycznych.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Hazard JB, Hawk WA, Crile G Jr: Medullary (solid) carcinoma of the thyroid – a clinicopathology entity. J Clin Endocrinol Metab 1959, 19: 152-61.
2. Eng C: RET proto-oncogene in the development of human cancer. J Clin Oncol 1999, 17 (1): 380-93.
3. Wiench M, et al.: Genetic diagnosis of multiple endocrine neoplasia type 2B. Endokrynologia Polska 2000, 51: 67-76.
4. Barbot N, et al.: Pentagastrin stimulation test and early diagnosis of medullary thyroid carcinoma using immunoradiometric assay of calcitonin: comparison with genetic screening in hereditary medullary thyroid carcinoma. J Clin Endocrinol Metab, 1994, 78: 114-20.
5. Donis-Keller H, et al.: Mutations of the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 1993, 2: 851-6.
6. Hofstra RM, et al.: A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 1994, 367: 375-6.
7. Mulligan LM, et al.: Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993, 363: 458-60.
8. Eng C, et al.: The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET Mutation Consortium analysis. JAMA 1996, 276: 1575-9.
9. Eng C, Mulligan LM: Mutations of the RET proto-oncogene in the multiple endocrine neoplasia type 2 syndromes, related sporadic tumors, and Hirschsprung disease. Human Mutation 1997, 9: 97-109.
10. Gagel RF, Cote GJ: Pathogenesis of medullary thyroid carcinoma. Thyroid Cancer. Kluwer Academic Publisher. Boston/Dordrecht/London, 1998.
11. Brandi ML, et al.: Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001, 86: 5658-71.
12. Gimm O, Sutter T, Dralle H: Diagnosis and therapy of sporadic and familial medullary thyroid carcinoma. J Cancer Res Clin Oncol 2001, 127: 156-65.
13. Kitamura Y, et al.: Novel germline RET protooncogene mutations associated with medullary thyroid carcinoma (MTC): mutation analysis in Japanese patients with MTC. Oncogene 1997, 14: 3103-6.
14. Berndt I, et al.: A new hot spot for mutations in the RET protooncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab 1998, 83: 770-4.
15. Bolino A, et al.: RET mutations in exons 13 and 14 of FMTC patients. Oncogene 1995, 10: 2415-9.
16. Eng C, et al.: AJ novel point mutation in the tyrosine kinase domain of the RET proto-oncogene in sporadic medullary thyroid carcinoma and in family with FMTC. Oncogene 1995, 10: 509-13.
17. Hofstra RM, et al.: A novel point mutation in the intracellular domain of the RET protooncogene in a family with medullary thyroid carcinoma. J Clin Endocrinol Metab 1997, 82: 4176-8.
18. Wohllk N, et al.: Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 1996, 81: 3740-5.
19. Jarząb B, et al.: Wczesna diagnostyka zespołu mnogiej gruczolakowatości wewnątrzwydzielniczej typu 2 poprzez analizę genetyczną germinalnych mutacji protoonkogenu RET. Endokrynol Pol 1999, 50: 127-34.
20. Lips CJ, et al.: Clinical screening as compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N Eng J Med 1994, 331: 828-35.
21. Włoch J, et al.: Profilaktyczne całkowite wycięcie tarczycy u nosicieli mutacji w protoonkogenie RET powodujących dziedzicznego raka rdzeniastego tarczycy. Pol Przegl Chir 2001, 73: 569-85.
22. Krassowski J, et al.: Oznaczanie kalcytoniny w rozpoznawaniu i ocenie wyników leczenia raka rdzeniastego tarczycy. Pol Tyg Lek 1989, 44: 757-9.
23. Pacini F, et al.: Routine measurement of serum calcitonin in nodular thyroid diseases allows the preoperative diagnosis of unsuspected sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 1994, 78: 826-9.
24. Wasylewski A, et al.: Przydatność oznaczania kalcytoniny w ocenie doszczętności zabiegu operacyjnego u chorych z rakiem rdzeniastym tarczycy. Endokrynol Pol 1981, 32: 239-44.
25. Januszewicz A: Nadciśnienie tętnicze. Zarys patogenezy, diagnostyki i leczenia. Medycyna Praktyczna 2002.
26. Rekomendacje Diagnostyka i Leczenie raka tarczycy przyjęte podczas III Konferencji naukowej „Rak tarczycy”, Szczyrk, 25.03.2006 Endokrynol Pol 2006, 57: 458-77.
27. Januszewicz A, i wsp.: Wytyczne dotyczące diagnostyki i leczenia chorych z guzem chromochłonnym. Nadciśnienie tętnicze 2006; 10: 1-19.
28. Włoch J: Postępowanie chirurgiczne w dziedzicznym raku rdzeniastym tarczycy: modyfikacje wynikające z diagnostyki mutacji germinalnych protoonkogenu RET i badania profilu molekularnego guzów (rozprawa habilitacyjna). Nowotwory (w druku).
29. Dralle H, et al.: Prophylactic thyreoidectomy in 75 children an adolescent with hereditary medullary thyroid carcinoma: German and Austrian experience. World J Surg 1998, 22: 744-750.
30. Skinner M, et al.: Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med 2005, 15; 353: 1105-13.
31. Baudin E, Travagli JP, Schlumberger M: How effective is prophylactic thyroidectomy in asymptomatic multiple endocrine neoplasia type 2A? Nat Clin Pract Endocrinol Metab 2006, 2: 256-7.
