© Borgis - Postępy Nauk Medycznych 7/2008, s. 463-471
*Andrzej Pławski1, Marta Podralska1, Piotr Krokowicz4, Jacek Paszkowski2, Jan Lubiński3, Ryszard Słomski1, 5
Rodzinna polipowatość gruczolakowata jelita grubego
Familial adenomatous polyposis of colon
1Instytut Genetyki Człowieka PAN w Poznaniu
Dyrektor Instytutu: prof. dr hab. Jerzy Nowak
2Katedra i Klinika Chirurgii Ogólnej, Gastroenterologii i Endokrynologii, Uniwersytet Medyczny w Poznaniu
Kierownik Kliniki: prof. dr hab. Michał Drews
3Zakład Genetyki i Patomorfologii, Pomorska Akademia Medyczna w Szczecinie
Kierownik Zakładu: prof. dr hab. med. Jan Lubiński
4 Katedra i Klinika Chirurgii Ogólnej i Endokrynologicznej Uniwersytet Medyczny w Poznaniu
Kierownik Katedry: prof. UM dr hab. Piotr Krokowicz
5 Katedra Biochemii i Biotechnologii, Uniwersytet Przyrodniczy w Poznaniu
Kierownik Katedry: prof. dr hab. Ryszard Słomski
Streszczenie
Występowanie licznych polipów w jelicie grubym i odbytnicy jest warunkowane predyspozycjami genetycznymi. Najczęstszą z nich jest rodzinna polipowatość jelita grubego (FAP), charakteryzująca się występowaniem licznych polipów w jelicie grubym i odbytnicy. Nieleczone polipy rozwijają się do nowotworu złośliwego, średnio w wieku 40 lat. FAP jest związany z występowaniem mutacji w genie supresorowym nowotworów APC, położonym na chromosomie 5q21. Gen APC koduje białko o masie 300kDa, które uczestniczy w kontroli proliferacji komórek śluzówki jelita poprzez degradację β-kateniny. Drugą z dziedzicznych predyspozycji do występowania nowotworów jelita grubego jest polipowatość związana z genem MUTYH (MAP). MAP jest również dziedziczną predyspozycją do występowania nowotworów, jednak liczba polipów jest mniejsza niż w przypadkach klasycznego FAP. Gen MUTYH jest zaangażowany w naprawę oksydacyjnych uszkodzeń DNA. FAP i MAP stanowią podłoże 1% wszystkich dziedzicznych predyspozycji do występowania raków jelita grubego. Wysokie ryzyko wystąpienia raka w obu chorobach sprawia, że należą do grupy bardzo istotnych zagadnień medycznych. W Polsce badania molekularne polipowatości jelit prowadzone są od ponad dziesięciu lat. W Instytucie Genetyki Człowieka PAN w Poznaniu utworzono bank DNA dla z polskich chorych z polipowatościami jelita. W banku DNA zgromadzono DNA od ponad 400 rodzin. Instytut Genetyki Człowieka w Poznaniu współpracuje z klinikami chirurgii i gastroenterologii oraz ośrodkami poradnictwa genetycznego na terenie całego kraju.
Summary
The occurrence of the numerous polyps in colon and rectum is conditioned by genetic predispositions. The most frequent is familial adenomatous polyposis (FAP), an autosomal dominant heritable cancer predisposition characterized by presence of numerous adenomatous polyps in the colon and rectum. The polyps progress to malignancy in untreated carriers of mutant gene with a median age at diagnosis of cancer of 40 years. FAP is linked with mutations of the APC tumor suppressor gene localized on chromosome 5q21. The APC gene encodes a 300kDa protein involved in cell proliferation control by degradation of the β-cathenine. The second predisposition is MUTYH associated polyposis (MAP). The MAP is a hereditary predisposition characterized by occurrence of polyps in colon but the number of polyps is lower than in classical FAP cases. The MUTYH gene is involved in the repairing of the DNA oxidative damages. FAP and MAP constitute the basis for about 1% of all colon cancers. The high risk of cancer observed in those syndromes make them important medical issues. The molecular studies of the polyposis of colon in Poland are performed for over ten years. In Institute of Human Genetics in Poznan the DNA Bank for Polish FAP patients was established. In the DNA Bank the DNA from over four hundreds families were collected. The Institute of Human Genetics in Poznan cooperates with surgery, gastroenterology clinics and genetics counseling centers from whole country.

Rodzinna polipowatość gruczolakowata (FAP, ang. familial adenomatous polyposis) stanowi około 1% wszystkich raków jelita grubego. De novo choroba występuje z częstością 1 na 8000 urodzeń. Wiek występowania objawów u chorych jest dość zróżnicowany, obserwuje się również różnice w wieku występowania objawów u rodzeństwa. Jednak można przyjąć, że wystąpienie raka jelita grubego w młodym wieku powinno być sygnałem do przeprowadzenia wywiadu rodzinnego, który pozwala na określenie, czy w rodzinie występuje wysoka dziedziczna predyspozycja. Występowanie pojedynczego przypadku choroby nie wyklucza wysokiej dziedzicznej predyspozycji, ponieważ chory może być pierwszym nosicielem mutacji. Objawy FAP występują wcześniej, niż w HNPCC i pojawiają się w drugiej dekadzie życia, choć obserwuje się przypadki występowania choroby nawet w wieku 5 lat. Podłożem genetycznym występowania polipów gruczolakowatych jest występowanie mutacji w genie APC w przypadku FAP i MUTYH w przypadku recesywnej formy polipowatości jelita.
Geny supresorowe nowotworów zaangażowane są w kontrolę proliferacji komórek. Produkty białkowe genów supresorowych biorą udział w kontroli cyklu komórkowego jako jego inhibitory, są także składnikami układu kontaktowego hamowania wzrostu. Geny supresorowe pełnią funkcję w utrzymywaniu liczby komórek na stałym poziomie. Zaburzenia tych mechanizmów prowadzą do zwiększenia częstości podziałów komórkowych, jak również wzrostu liczby błędów powstających podczas podziału. Prowadzi to do gromadzenia zmian w materiale genetycznym i wyselekcjonowania nieśmiertelnego klonu o bardzo częstych podziałach komórkowych, zdolnego zasiedlać inne tkanki.
Mutacje genów supresorowych mają charakter recesywny, ponieważ w przypadku genów supresorowych fenotyp mutacji jest maskowany przez prawidłowy allel genu. W inicjacji procesu nowotworowego występują dwie niezależne mutacje w obrębie locus genu supresorowego. W przypadku nosicielstwa zmutowanego allelu genu APC ryzyko wystąpienia drugiej mutacji, a tym samym inicjacji choroby nowotworowej, jest bardzo wysokie.
Historia badań genu APC
FAP została zidentyfikowana jako dziedziczny zespół chorobowy już w latach 20-tych dwudziestego wieku. W 1972 r. został opisany zespół Gardnera, który jest postacią FAP charakteryzującą się nie tylko obecnością setek czy tysięcy polipów w jelicie, lecz także kostniaków oraz przebarwień siatkówki (CHRPE, ang. congenital hypertrophy of the retinal pigment epithelium). Występowanie FAP zaczęto wiązać z regionem q21-q22 chromosomu 5 na podstawie obserwacji dużej delecji wykrytej w badaniach cytogenetycznych oraz wyników badań sprzężeń markerów RFLP pacjenta z zespołem Gardnera i zaawansowanym rozwojem polipów w jelicie grubym. Pod koniec lat osiemdziesiątych badania sprzężeń ukierunkowały poszukiwania genu na region obejmujący oddalone od siebie o 150 kpz geny APC i MCC. W 1991 r. u chorych z FAP przebadano trzy geny: DP1, SRP19 i DP2.5 znajdujące się w regionie, który uległ delecji. U dwóch pacjentów z FAP zaobserwowano 4 mutacje w genie DP2.5 (obecnie gen APC) prowadzące do powstania kodonu Stop, z których jedna była przekazana potomstwu. W następnym roku przebadano 79 chorych z FAP i zaobserwowano mutacje w genie APC u 67% chorych. W 92% były to mutacje prowadzące do skrócenia produktu białkowego genu APC. W wielu krajach zaczęto badać występowanie mutacji w genie APC i utworzono bazę danych mutacji dziedzicznych i somatycznych, w których zgromadzono dane o 826 mutacjach dziedzicznych i 650 mutacjach somatycznych (8). Funkcja białka APC była badana od 1993 r., gdy zaobserwowano, że wiąże się ono z β-kateniną, co wskazywało na uczestniczenie w adhezji komórek. W naszym kraju Bank DNA chorych, z FAP został utworzony w 1997 roku w Instytucie Genetyki Człowieka PAN w Poznaniu z inicjatywy A. Pławskiego i R. Słomskiego.
Budowa genu APC i funkcja białka APC
Gen APC (ang. adenomatous polyposis coli) położony jest na chromosomie 5 w regionie q21 i zawiera 21 eksonów. Charakterystyczną cechą genu APC jest występowanie dużego eksonu 15, który obejmuje ponad 70% sekwencji kodującej. Ekspresja genu obserwowana jest we wszystkich tkankach. Produktem transkrypcji jest mRNA o długości 8538 nukleotydów. Pierwsze eksony genu mogą tworzyć specyficzne tkankowo alternatywne transkrypty, np. w mózgu występuje produkt genu APC, dla którego kodon Start położony jest w eksonie BS (ang. brain specific). Wyeliminowanie kodonu 1, który koduje domenę super spirali, o której wiadomo, że służy do homo- lub heterodimeryzacji, wpływa na funkcjonalność produktu białkowego. Alternatywne składanie początkowych eksonów genu może być związane z występowaniem łagodnej formy polipowatości – AAPC (ang. attenuated adenomatous polyposis coli). Podobny efekt zaobserwowano w jednej z dwóch rodzin z mutacjami na końcu 3´ eksonu 9, gdzie w jednym przypadku wykazano modyfikujący efekt alternatywnego składania, prowadzący do łagodnej formy FAP. W związku z tym uważa się, że rodzaj, położenie mutacji jak również efekt alternatywnego składania wpływają na przebieg choroby.
Bardzo ciekawa jest obserwacja międzygenowego składania, w którego wyniku z genu APC zostaje usunięty ekson 14 i obejmujący prawie 70% genu ekson 15, a pozostały fragment łączy się z końcem 3´ genu SRP1. Wycięcie eksonu 14 lub 15 prowadzi do powstania dwóch izoform produktu różniących się zdolnością wiązania mikrotubul i β-kateniny, jak również sekwencją regionu 3´ niepodlegającego translacji, co może wpływać na stabilność mRNA oraz na funkcje produktu. Alternatywne składanie genu jest w tym przypadku związane z regulacją aktywności białka APC i nasuwa przypuszczenie, że pełni ono wiele różnych funkcji w komórce zwłaszcza, że w alternatywnym składaniu bierze udział ponad 75% eksonów.
Pełnej długości białko APC składa się z 2843 aminokwasów i występuje w cytoplazmie oraz w jądrze komórkowym. Dotychczas poznano kilka białek wchodzących w interakcję z białkiem APC. Należą do nich białko DLG, białko mikrotubul, GSKβ-3, β-katenina, γ-katenina, p34, Tid56, białko Auxin. Interakcje z wieloma białkami oznaczają, że białko APC bierze udział w regulacji wielu procesów w komórce, obejmujących podział, migrację, adhezję i różnicowanie komórek (ang. cell fate determination). W białku APC wyodrębniono kilka funkcjonalnych domen. Domena zasadowa obejmuje aminokwasy 2200-2400 (1). Fragment końcowy białka, między aminokwasami 1-171, zaangażowany jest w oligomeryzację. W białku APC występują dwa miejsca wiązania β-kateniny – we fragmencie obejmującym trzy 15-nukleotydowe powtórzenia między aminokwasami 1020-1169 i w regionie 20 aminokwasowych powtórzeń między aminokwasami 1324-2075. Wiązanie z mikrotubulami, występujące podczas zwiększonej ekspresji genu, odbywa się za pomocą fragmentu obejmującego aminokwasy 2130-2483. Aminokwasy 2560-2843 są miejscem wiązania z białkiem EB1, a aminokwasy 2771-2843 wiążą się z białkiem DLG. Nie wyodrębniono regionu związanego z procesem apoptozy, zaobserwowano jednak, że ekspresja prawidłowego białka APC w komórkach linii nowotworowej jelita prowadzi do wystąpienia tego zjawiska. W komórkach śluzówki jelita grubego produkt genu APC o masie 300 kDa uczestniczy w kontaktowym hamowaniu wzrostu komórek.
Białko APC oddziałuje także z γ-kateniną i β-kateniną. Obydwa białka wiążą się z białkiem adhezyjnym E-kadheryną. Fearon (1997) zaproponował schemat, w którym białko APC bierze udział w transdukcji sygnału i przez degradację β-kateniny wpływa na aktywność czynnika transkrypcji Tcf4 (ang. T-cell transcription factor 4) (ryc. 1). Białkiem regulującym tworzenie kompleksu białka APC z β-kateniną jest kinaza białkowa ZW3/GSK3β. Fosforylacja białka APC aktywuje wiązanie β-kateniny. Aktywność kinazy GSK3β jest regulowana poprzez białko dsh, wchodzące w interakcje z produktem genu WNT1. Białko APC związane z kinazą ZW3/GSK3β posiada zdolność inhibowania transkrypcji indukowanej przez β-kateninę. W przypadku utraty funkcji produktu genu APC następuje aktywacja czynnika transkrypcji Tcf4 ( TCF7L2). Komórka jest pobudzona do proliferacji w wyniku aktywacji transkrypcji genu c-MYC przez Tcf4. Produkt genu c-MYC występuje w jądrze komórkowym i posiada zdolność wiązania z DNA, aktywuje gen wzrostu – dekarboksylazę ornityny ( ODC1) i gen CDC25A, ponadto jest inhibitorem genu GAS1. Wykazano również, że aktywowany kompleks β-katenina-Tcf4 indukuje ekspresję Tcf1. W komórkach śluzówki jelita grubego gen APC działa jako regulator cyklu komórkowego inhibujący przez regulacje poziomu β-kateniny sygnał proliferacji pochodzący od transmembranowego białka E-kadheryny. W przypadku utraty funkcjonalności tego genu w tkance zostaje zaburzona równowaga między podziałami i śmiercią komórek. Białko APC oddziałuje także z p34, Tid56 i białkiem Auxin.
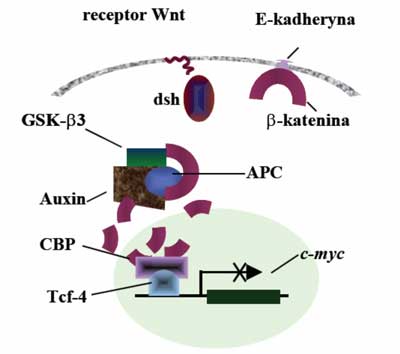
Ryc. 1. Funkcja białka APC w kontroli proliferacji komórek.
Mutacje genu APC
Mutacjami genu APC są najczęściej delecje lub insercje kilku par zasad, rzadziej substytucje. Większość mutacji (98%) prowadzi do skrócenia produktu białkowego. W przypadku substytucji, które stanowią 30% mutacji, kodon Stop powstaje w miejscu mutacji, a w przypadku delecji lub insercji stanowiących 68% mutacji powstaje w jej pobliżu w wyniku zmiany ramki odczytu. W genie występuje region o podwyższonej częstości występowania mutacji MCR (ang. mutation cluster region), który obejmuje kodony 1055-1309. W regionie tym występuje 23% wszystkich mutacji germinalnych. Ponadto wśród mutacji germinalnych zaobserwowano podwyższoną częstość trzech mutacji: delecji 5 pz w kodonie 1309 (10%), delecji 5 pz w kodonie 1061 oraz delecji 4 pz w kodonie 1064. Większość mutacji występuje w regionie 5´ eksonu 15 genu APC. Charakterystyczną cechą FAP jest utrata heterozygotyczności (LOH, ang. loss of heterozygosity) w genie APC powstająca w wyniku mutacji somatycznej w drugim allelu genu APC. Rozkład częstości tych mutacji jest różny od mutacji germinalnych. Do 60% mutacji somatycznych znajduje się we fragmencie, który obejmuje 8% genu między kodonami 1286 i 1513. Mutacje somatyczne występują w dwóch gorących miejscach (ang. hot spots) w kodonie 1309 i kodonie 1450. Mutacje w genie APC występują również w przypadkach raków jelita grubego niezwiązanych z polipowatością. W przypadkach tych nowotworów występują pojedyncze guzy, ponieważ musi dojść do mutacji w jednym z alleli genu APC, a następnie utraty heterozygotyczności w wyniku mutacji somatycznych. W przypadku zespołu Lynch (HNPCC) proces ten jest przyspieszany przez dziedziczone mutacje w genach naprawy DNA. Według najnowszych badań, dla inicjacji nowotworu jelita grubego niezwiązanego, z FAP nie musi wystąpić LOH w APC. W połowie sporadycznych nowotworów, w których nie zaobserwowano zmian w APC, powstanie nowotworu związane jest z heterozygotycznymi mutacjami w genie β-kateniny ( CTNNB1). Mutacje występują w miejscu fosforylacji β-kateniny przez kinazę GSK 3β. Powodują one wyłączenie regulacyjnej funkcji genu APC, co prowadzi do kumulacji β-kateniny, a tym samym ekspresji genu MYC i rozwoju nowotworu jelita grubego. β-katenina znajduje się za genem APC (ang. downstream) w szlaku kontaktowego hamowania wzrostu i w przypadku mutacji w tym genie proces nowotworowy przebiega niezależnie od stanu genu APC.
Podjęto próbę określenia związku między położeniem mutacji terminujących translację a obrazem klinicznym choroby i wystąpieniem objawów pozajelitowych. Zaobserwowano, że wystąpienie kodonu Stop przed kodonem 157 powoduje zmniejszenie liczby polipów i łagodny przebieg choroby. Klasyczny przebieg polipowatości z licznymi polipami ma miejsce w przypadku wystąpienia sygnału Stop między kodonami 169 a 1600. Mutacje występujące między kodonami 1403 i 1587 prowadzą do nasilenia objawów pozajelitowych. Występowanie włókniakowatości naciekowej związane jest z mutacjami w regionie między kodonami 1445 a 1578. Mutacje w końcu 3´ genu APC dają zróżnicowany obraz przebiegu choroby zarówno pod względem liczby polipów, jak również występowania objawów pozajelitowych.
Progresja zmian morfologicznych i genetycznych w rodzinnej polipowatości gruczolakowatej
Dziedziczenie zmutowanego allelu genu APC nie wywołuje obrazu klinicznego choroby. Pojawienie się objawów klinicznych związane jest z sekwencją dalszych zdarzeń w komórce. Zarówno w przypadku FAP jak i HNPCC komórki charakteryzują się bardzo wysokim ryzykiem wystąpienia utraty hetrozygotyczności w locus genu APC. W czasie rozwoju nowotworu zachodzą zmiany w obrębie genów supresorowych, onkogenów i genów naprawy DNA. Delecje obserwuje się w regionach występowania genów hamujących proliferację, położonych na chromosomach 5q ( APC, MCC), 17p ( TP53) i 18q ( DCC) oraz obserwuje się mutacje punktowe w obrębie protoonkogenu K-ras. W przypadku FAP ryzyko utraty heterozygotyczności jest bardzo wysokie, ponieważ jeden z alleli dziedziczony jest w formie zmutowanej. Ryzyko inicjacji nowotworu jest również wysokie w wyniku dziedziczenia mutacji. Mimo różnych przyczyn pierwszym etapem inicjacji choroby nowotworowej jest utrata heterozygotyczności w locus APC, prowadząca do zwiększenia proliferacji komórek w wyniku aktywacji transkrypcji protoonkogenu c-MYC. Częste podziały komórek prowadzą do wyselekcjonowania klonu z uszkodzonym genem MCC, co powoduje dysplazję i w dalszym etapie powstanie gruczolaka stopnia pierwszego. Wzrost tempa podziałów komórek powoduje dalsze gromadzenie błędów genetycznych i w wyniku następuje aktywacja protoonkogenu K-ras i delecja DCC (ang. deleted in colorectal cancer). Kolejnym etapem rozwoju nowotworu jest utrata funkcjonalności produktu genu TP53, co prowadzi do powstania gruczolakoraka, a dalsze gromadzenie błędów do powstania nowotworu inwazyjnego. Różnica w podłożu molekularnym FAP i HNPCC sprawia, że pojedyncze guzy w HNPCC rozwijają się szybciej do formy inwazyjnej niż w przypadku FAP.
Fenotyp mutacji w genie APC
Rodzinna polipowatość gruczolakowata (FAP) (OMIM 175100)
W około 60-85% przypadków FAP wykrywane są mutacje w genie APC. Objawy w postaci licznych (setki lub tysiące) polipów w śluzówce jelita grubego pojawiają się pod koniec drugiej dekady życia. Polipy charakteryzują się znacznym potencjałem do rozwoju w kierunku nowotworu złośliwego. Transformacja polipów o charakterze gruczolaków w nowotwór złośliwy występuje średnio w wieku 40 lat, ale może wystąpić w okresie od późnego dzieciństwa do 70 lat. Pierwszymi objawami wystąpienia polipów są biegunki i krew w stolcu. Najwcześniejsze wystąpienie objawów polipowatości zaobserwowano w wieku 5 lat. Wraz z objawami w jelicie grubym obserwuje się występowanie jednego lub kilku objawów pozajelitowych, do których zalicza się zmiany w wyższych odcinkach przewodu pokarmowego, zmiany w siatkówce oka, nowotwory poza jelitem grubym, zmiany skórne i zmiany w kośćcu. W wyższych odcinkach przewodu pokarmowego najczęściej występują gruczolaki żołądka, gruczolaki dwunastnicy i hypertroficzne polipy dna żołądka. Gruczolaki żołądka występują częściej niż gruczolaki dwunastnicy. Poza tym obserwuje się występowanie wątrobiaków, raków brodawkowych tarczycy (wśród pacjentów z tym nowotworem dominują kobiety – 94%) oraz nowotwory nadnerczy. Kolejnym typem nowotworu występującego w FAP jest włókniakowatość naciekowa (ang. desmoid tumors), jej pojawienie następuje częściej po zabiegu chirurgicznym. Choroba ta występuje rzadziej u mężczyzn z FAP (8%) niż u kobiet (13%). Przebadano związek lokalizacji mutacji skracającej produkt genu APC, z występowaniem włókniakowatości naciekowej. Występowanie włókniakowatości naciekowej odnotowano u wszystkich pacjentów z FAP, u których mutacja wystąpiła między kodonami 1445 a 1578.
Zespół Gardnera (OMIM 175100.0006) jest formą FAP z licznymi objawami pozajelitowymi w formie CHRPE, torbieli gruczołów łojowych skóry, kostniaków i włókniakowatości naciekowej, ze zmianami w uzębieniu polegającymi na zmianach liczby zębów oraz występowaniu długich i zaostrzonych korzeni zębowych. Występowanie CHRPE wiązane jest z położeniem mutacji w genie APC. Przebarwienie siatkówki nie występuje, jeżeli produkt zmutowanego genu jest mniejszy niż 50 kDa, a ekson 9 uważany jest za graniczny między mutacjami niepowodującymi, a powodującymi przebarwienie. W regionie 3´ genu APC granicą występowania przebarwienia jest kodon 1387. Długość produktu ma wpływ na obraz przebarwienia. Produkty białkowe genu o długości poniżej 1014 aminokwasów przedstawiają obraz małego okrągłego przebarwienia (ang. small round pigmented) lub dużego okrągłego przebarwienia (ang. large round pigmented). Produkty białkowe powyżej 1014 aminokwasów powodują wzrost występowania pozostałych dwóch typów zmian w siatkówce dna oka tj. owalnego przebarwienia okrążonego aureolą (ang. oval pigmented with a surrounding halo) i dużego okrągłego odbarwienia (ang. large round de-pigmented). Kodon 1014 jest miejscem wiązania dwóch białek cytoplazmatycznych α i β-kateniny; są one istotne dla białka czynnego w adhezji komórek E-kadheryny. Produkty dłuższe od 1014 aminokwasów mogą wiązać się z tymi białkami, co może wpływać na zmianę fenotypu przebarwienia. Przebarwienie barwnikowe siatkówki jest stosowane jako marker nosicielstwa zmutowanego allelu, ale rozwój technik biologii molekularnej, jak i występowanie tylko u części rodzin ograniczyły jego znaczenie.
Łagodna forma rodzinnej polipowatości gruczolakowatej jelita grubego (AAPC)
Łagodna forma rodzinnej polipowatości jelita grubego AAPC (ang. attenuated adenomatous polyposis coli) charakteryzuje się występowaniem małej liczby polipów od kilku do stu. Z objawów pozajelitowych rzadko obserwuje się występowanie polipów dna żołądka. Występowanie tej formy rodzinnej polipowatości jelita związane jest z występowaniem mutacji na końcu 5´ genu APC. Przyjmuje się, że kodon 159 jest granicą między mutacjami powodującymi AAPC a FAP, jednak ustalenie jednoznacznej granicy jest trudne, ponieważ ta forma choroby wystąpiła również w przypadkach, gdy mutacja prowadziła do powstania kodonu Stop w eksonie 9 genu APC.
Polipowatość recesywna (MIM 608456) MAP (ang. MUTYH associated polyposis)
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H, Joslyn G, Stevens J, Spirio L, Robertson M, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991, 66: 589-600.
2. Bisgaard ML, Fenger K, Bülow S, Niebuhr E, Mohr J. Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994, 3 (2): 121-5.
3. Lynch HT, Smyrk TC. Identyfying Hereditary Nonpolyposis Colorectal Cancer. N Engl J Med 1998, 338: 1537-8.
4. Distante S, Nasioulas S, Somers GR, Cameron DJ, Young MA, Forrest SM, Gardner RJ. Familial adenomatous polyposis in a 5 year old child: a clinical, pathological, and molecular genetic study. J Med Genet 1996, 33: 157-60.
5. Knudson AJR. Mutation and cancer: statistical study of retinoblastoma. Proc Nat Acad Sci 1971, 68: 820-3.
6. Herrera L, Kakati S, Gibas L, Pietrzak E, Sandberg AA. Gardner syndrome in a man with an interstitial deletion of 5q. Am J Med Genet 1986, 25: 473-6.
7. Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1992, 1: 229-33.
8. Rubinfeld B, Souza B, Albert I, Müller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with beta-catenin. Science 1993, 262: 1731-4.
9. Santoro IM, Groden J. Alternative splicing of the APC gene and its association with terminal differentiation. Cancer Res 1997, 57: 488-94.
10. Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB, Smith KJ, Preisinger AC, Hedge P, McKechnie D et al. Identification of FAP locus genes from chromosome 5q21. Science 1991, 253: 661-5.
11. Lambertz S, Ballhausen WG. Identification of an alternative 5´ untranslated region of the adenomatous polyposis coli gene. Hum Genet 1993, 90: 650-2.
12. Samowitz WS, Thliveris A, Spirio LN, White R. Alternatively spliced adenomatous polyposis coli (APC) gene transcripts that delete exons mutated in attenuated APC. Cancer Res 1995, 55: 3732-4.
13. Sulekova Z, Reina-Sanchez J, Ballhausen WG. Multiple APC messenger RNA isoforms encoding exon 15 short open reading frames are expressed in the context of a novel exon 10A-derived sequence. Int J Cancer 1995, 63: 435-41.
14. Fearon ER. Human cancer syndromes: clues to the origin and nature of cancer. Science 1997, 278: 1043-50.
15. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science 1998, 281: 1509-12.
16. Roose J, Huls G, van Beest M, Moerer P, van der Horn K, Goldschmeding R, Logtenberg T, Clevers H. Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science 1999, 285: 1923-6.
17. Mandl M, Paffenholz R, Friedl W, Caspari R, Sengteller M, Propping P. Frequency of common and novel inactivating APC mutations in 202 families with familial adenomatous polyposis. Hum Mol Genet 1994, 3: 181-4.
18. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990, 61: 759-67.
19. Morin PJ. beta-catenin signaling and cancer. Bioessays 1999, 21: 1021-30.
20. van der Luijt RB, Khan PM, Vasen HF, Tops CM, van Leeuwen-Cornelisse IS, Wijnen JT, van der Klift HM, Plug RJ, Griffioen G, Fodde R. Molecular analysis of the APC gene in 105 Dutch kindreds with familial adenomatous polyposis: 67 germline mutations identified by DGGE, PTT, and southern analysis. Hum Mutat 1997, 9: 7-16.
21. Klemmer S, Pascoe L, DeCosse J. Occurrence of desmoids in patients with familial adenomatous polyposis of the colon. Am J Med Genet 1987, 28: 385-92.
22. Bishop DT, Thomas HJ. The genetics of colorectal cancer. Cancer Surv 1990, 9: 585-604.
23. Wallis YL, Macdonald F, Hultén M, Morton JE, McKeown CM, Neoptolemos JP, Keighley M, Morton DG. Genotype-phenotype correlation between position of constitutional APC gene mutation and CHRPE expression in familial adenomatous polyposis. Hum Genet 1994, 94: 543-8.
24. Soravia C, Berk T, Madlensky L, Mitri A, Cheng H, Gallinger S, Cohen Z, Bapat B. Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet 1998, 62: 1290-301.
25. Sampson JR, Dolwani S, Jones S, Eccles D, Ellis A, Evans DG, Frayling I, Jordan S, Maher ER, Mak T, Maynard J, Pigatto F, Shaw J, Cheadle JP. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003, 362: 39-41.
26. Farrington SM, Tenesa A, Barnetson R, Wiltshire A, Prendergast J, Porteous M, Campbell H, Dunlop MG. Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am J Hum Genet 2005, 77: 112-9.
27. Barnetson RA, Devlin L, Miller J, Farrington SM, Slater S, Drake AC, Campbell H, Dunlop MG, Porteous ME. Germline mutation prevalence in the base excision repair gene, MYH, in patients with endometrial cancer. Clin Genet 2007, 72: 551-5.
28. Nielsen M, Hes FJ, Nagengast FM, Weiss MM, Mathus-Vliegen EM, Morreau H, Breuning MH, Wijnen JT, Tops CM, Vasen HF. Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis. Clin Genet 2007, 71: 427-33.
29. Mastronardi L, Ferrante L, Lunardi P, Cervoni L, Fortuna A. Association between neuroepithelial tumor and multiple intestinal polyposis (Turcot´s syndrome): report of a case and critical analysis of the literature. Neurosurgery 1991, 28: 449-52.
30. Drews M, Krokowicz P, Banasiewicz T. Diagnostyka, profilaktyka i leczenie chorych z polipowatością rodzinną jelita grubego. Nowiny Lekarskie 1999, 68: 725-34.
31. Spigelman AD, Murday V, Phillips RK. Cancer and the Peutz-Jeghers syndrome. Gut 1989, 30: 1588-90.
32. Houlston R, Bevan S, Williams A, Young J, Dunlop M, Rozen P, Eng C, Markie D, Woodford-Richens K, Rodriguez-Bigas MA, Leggett B, Neale K, Phillips R, Sheridan E, Hodgson S, Iwama T, Eccles D, Bodmer W, Tomlinson I. Mutations in DPC4 (SMAD4) cause juvenile polyposis syndrome, but only account for a minority of cases. Hum Mol Genet 1998, 7: 1907-12.
33. Slupska MM, Baikalov C, Luther WM, Chiang JH, Wei YF, Miller JH. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J Bacteriol 1996, 178: 3885-92.
34. Sieber OM, Lipton L, Crabtree M, Heinimann K, Fidalgo P, Phillips RK, Bisgaard ML, Orntoft TF, Aaltonen LA, Hodgson SV, Thomas HJ, Tomlinson IP. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med 2003, 348: 791-9.
35. Al-Tassan N, Eisen T, Maynard J, Bridle H, Shah B, Fleischmann C, Sampson JR, Cheadle JP, Houlston RS. Inherited variants in MYH are unlikely to contribute to the risk of lung carcinoma. Hum Genet 2004; 114: 207-10.
36. Jones S, Emmerson P, Maynard J, Best JM, Jordan S, Williams GT, Sampson JR, Cheadle JP. Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C->T:A mutations. Hum Mol Genet 2002, 11: 2961-7.
37. Plawski A, Slomski R. Geny Supresorowe Nowotworów. Postępy biologii komórki 1998, 25: 251-264.
38. Plawski A, Nowakowska D, Podralska M, Lipinski D, Steffen J, Slomski R. The AAPC case, with an early onset of colorectal cancer. Int J Colorectal Dis 2007, 22: 449-51.
39. Brozek I, Plawski A, Podralska M, Kanka C, Slomski R, Limon J. Thyroid cancer in two siblings with FAP syndrome and APC mutation. Int J Colorectal Dis 2008, 23: 331-2.
40. Skrzypczak M et al. The MYH gene status in Polish FAP patients without the APC gene mutations. Hereditary Cancer in Clinical Practice 2006, 4: 43-7.
41. Kubinska I et al. Using of molecular sreening in children with familial polyposis background - own experience. Polish Journal of Environmental Studies 2006, 15: 98-101.
42. Plawski A, Lubiński J, Banasiewicz T, Paszkowski J, Lipinski D, Strembalska A, Kurzawski G, Byrski T, Zajaczek S, Hodorowicz-Zaniewska D, Gach T, Brozek I, Nowakowska D, Czkwaniec E, Krokowicz P, Drews M, Zeyland J, Juzwa W, Słomski R. Novel germline mutations in the adenomatous polyposis coli gene in Polish families with familial adenomatous polyposis. J Med Genet 2004, 41: E11.
43. Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996, 87: 803-9.
44. Chiu CH, McEntee MF, Whelan J. Sulindac causes rapid regression of preexisting tumors in Min/+ mice independent of prostaglandin biosynthesis. Cancer Res 1997, 57: 4267-73.
