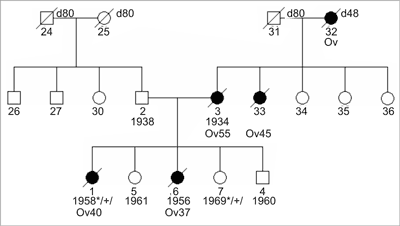
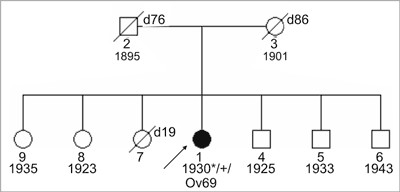
Familial breast cancer was first recognized in the Roman medical literature of 100 AD (1). The first documentation of familial clustering of breast cancer in modern times was published by Broca, who reported 10 cases of breast cancer in 4 generations of his wife’s family (2). In the middle of nineties it was proven at molecular level that substantial number of breast and ovarian cancers has hereditary monogenic etiology (3, 4). Evaluation of frequency of pedigree-clinical signs characteristic for strong aggregations of breast/ovarian cancers among consecutive cases of cancers of these organs as well as analyses of cancer incidence in monozygotic tweens indicate that about 30% of breast and ovarian cancers develop because of strong genetic predisposition (5). In other breast/ovarian cancers significance of genetic factors was underestimated. However, recently it was possible to show characteristic constitutional baground influencing development of cancer also in patients with sporadic neoplasms. Therefore now, scientists think that in almost all patients with cancer a certain genetic backgroung should be detectable although influencing cancer risk with different degree. Genetic abnormalities strongly related with cancer are called high risk changes (genes) and abnormalities influencing cancer development with lower degree are called moderate risk changes (genes). In Polish population most frequently strong genetic predisposition to breast/ovarian cancers are related to mutations in BRCA1, CHEK2 or PALB2 genes. Mutations in BRCA2 gene are observed relatively rare. Mutations in these genes most often apeare as syndromes of hereditary breast cancer – site specific (HBC-ss), hereditary breast-ovarian cancer (HBOC) and hereditary ovarian cancer (HOC). In family members of families with HBC-ss syndrome only breast cancers but not ovarian cancers are observed. In HBOC syndrome families with both – breast and ovarian cancers are diagnosed and in HOC syndrome only ovarian but not breast cancers are detected. Operational clinical-pedigree criteria which we use in order to diagnose the discussed syndromes are summarized in table 1. In vast majority of cancer cases related to moderate risk genes family history is negative. HBC-ss, HBOC, HOC syndromes are clinically and molecullary heterogenous. Mutations in BRCA1 and BRCA2 genes are the most frequent cause of these syndromes. Recently, it was shown that in substantial number of such families, the syndrome develop because of truncating mutations in CHEK2 gene or PALB2 genes (6, 7).
So far, several changes with potential significance in modification of the cancer risk have been identified. Multicenter study of CIMBA consortium suggest that these changes alone are weak and likely the effect is variable in different populations (14-29).
In Poland in about 30% of families with definitively diagnosed HBC-ss and HBOC syndromes and in about 40% of families with HOC syndrome, BRCA1 or BRCA2 mutations are not detected. In rare cases it is possible to diagnose one of rare syndromes listed in table 3. In these syndromes breast/ovarian cancers are observed with higher frequency. Many groups in the world try to identify new genes related to high breast cancer risk.
– carriers of mutations of high breast/ovarian cancer risk; usually around 50% of female family members shoud be included into program,
1. Lynch HT: Genetics and breast cancer. Van Nostrand-Reinhold, New York 1981.
2. Broca P: Traite de tumeurs. Paris, Asselin 1866.
3. Miki Y, Swensen J, Shattuck-Eidens D et al.: A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266: 66-71.
4. Wooster R, Bignell G, Lancaster J et al.: Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378: 789-792.
5. Lichtenstein P, Holm NV, Verkasalo PK et al.: Environmental and heritable factors in the causation of cancer – analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000; 343: 78-85.
6. Cybulski C, Wokołorczyk D, Jakubowska A et al.: Risk od Breast Cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol 2011 Aug 29; 29(28): 3747-3752.
7. Cybulski C, Lubiński J, Wokołorczyk D et al.: Mutations Predisposing to Breast Cancer in 12 Candidate Genes in Breast Cancer Patients from Poland. Clin Genet 2014 Oct 20. doi: 10.1111/cge.12524.
8. Antoniou AC, Pharoah PD, Narod S et al.: Breast and ovarian cancer risks to carriers of the BRCA1 5382insC and 185delAG and BRCA2 6174delT mutations: a combined analysis of 22 population based studies. J Med Genet 2005; 42: 602-603.
9. Gronwald J, Huzarski T, Byrski B et al.: Cancer risks in first degree relatives of BRCA1 mutation carriers: effects of mutation and proband disease status. J Med Genet 2006; 43: 424-428.
10. Risinger JI, Berchuck A, Kohler MF et al.: Mutations of the E-cadherin gene in human gynecologic cancers. Nat Genet 1994; 7: 98-102.
11. Thorlacius S, Struewing JP, Hartge P et al.: Population-based study of risk of breast cancer in carriers of BRCA2 mutation. Lancet 1998; 352: 1337-1339.
12. Metcalfe K, Lubinski J, Lynch HT et al.: Family history of cancer and cancer risks in women with BRCA1 or BRCA2 mutations. J Natl Cancer Inst 2010 Dec 15; 102(24): 1874-1878.
13. Lubinski J, Huzarski T, Byrski T et al.: The risk of breast cancer in women with a BRCA1 mutation from North America and Poland. Int J Cancer 2012 Jul 1; 131(1): 229-234.
14. Osorio A, Milne RL, Alonso R et al.: Evaluation of the XRCC1 gene as a phenotypic modifier in BRCA1/2 mutation carriers. Results from the Consortium of Investigators of Modifiers of BRCA1/BRCA2 (CIMBA). British Journal of Cancer 2011 Apr 12; 104(8): 1356-1361. Epub 2011 Mar 22.
15. Antoniou AC, Kartsonaki C, Sinilnikova OM et al.: Common alleles at 6q25.1 and 1p11.2 are associated with breast cancer risk for BRCA1 and BRCA2 mutation carriers. Human Molecular Genetics 2011 Aug 15; 20(16): 3304-3321. Epub 2011 May 18.
16. Antoniou AC, Wang X, Fredericksen ZS et al.: A locus on 19p13 modifies risk of breast cancer in BRCA1 mutation carriers and is associated with hormone receptor negative breast cancer in the general population. Nature Genetics 2010 Oct; 42(10): 885-892. Epub 2010 Sep 19.
17. Engel C, Versmold B, Wappenschmidt B et al.: Association of the variants CASP8 D302H and CASP10 V410I with breast and ovarian cancer risk in BRCA1 and BRCA2 mutation carriers. Cancer Epidemiology Biomarkers and Prevention 2010 Nov; 19(11): 2859-2868. Epub 2010 Oct 26.
18. Ramus SJ, Kartsonaki C, Gayther SA et al.: Genetic variation at 9p22.2 and ovarian cancer risks in BRCA1 and BRCA2 mutation carriers. Journal of the National Cancer Institute 2011 Jan 19; 103(2): 105-116. Epub 2010 Dec 17.
19. Antoniou AC, Beesley J, McGuffog L et al.: Common breast cancer susceptibility alleles and the risk of breast cancer for BRCA1 and BRCA2 mutation carriers: implications for risk prediction. Cancer Research 2010 Dec 1; 70(23): 9742-9754. Epub 2010 Nov 30.
20. Gaudet MM, Kirchhoff T, Green T et al.: Common genetic variants and modification of penetrance of BRCA2-associated breast cancer. PLoS Genet 2010 Oct 28; 6(10): e1001183.
21. Walker LC, Fredericksen ZS, Wang X et al.: Evidence for SMAD3 as a modifier of breast cancer risk in BRCA2 mutation carriers. Breast Cancer Research 2010; 12(6): R102. Epub 2010 Nov 29.
22. Wang X, Pankratz VS, Fredericksen Z et al.: Common variants associated with breast cancer in genome-wide association studies are modifiers of breast cancer risk in BRCA1 and BRCA2 mutation carriers. Hum Mol Genet 2010 Jul 15; 19(14): 2886-2897. Epub 2010 Apr 23.
23. Osorio A, Milne RL, Pita G et al.: Evaluation of a candidate breast cancer associated mSNP in ERCC4 as a risk modifier in BRCA1 and BRCA2 mutation carriers. Results from the consortium of investigators of modifiers of BRCA1/BRCA2 (CIMBA). British Journal of Cancer 2009 Dec 15; 101(12): 2048-2054. Epub 2009 Nov 17.
24. Jakubowska A, Rozkrut D, Antoniou A et al.: The Leu33Pro polymorphism in the ITGB3 gene does not modify BRCA1/2-associated breast or ovarian cancer risks: results from a multicenter study among 15 542 BRCA1 and BRCA2 mutation carriers. Breast Cancer Research and Treatment 2010 Jun; 121(3): 639-649. Epub 2009 Oct 30.
25. Antoniou AC, Sinilnikova OM, McGuffog L et al.: Common variants in LSP1, 2q35 and 8q24 and breast cancer risk for BRCA1 and BRCA2 mutation carriers. Human Molecular Genetics 2009 Nov 15; 18(22): 4442-4456. Epub 2009 Aug 5.
26. Osorio A, Pollán M, Pita G et al.: An evaluation of the polymorphisms Ins16bp and Arg72Pro in p53 as breast cancer risk modifiers in BRCA1 and BRCA2 mutation carriers. British Journal of Cancer 2008 Sep 16; 99(6): 974-977.
27. Antoniou AC, Spurdle AB, Sinilnikova OM et al.: Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCA1 and BRCA2 mutation carriers. Am J Hum Genet 2008 Apr; 82(4): 937-948. Epub 2008 Mar 20.
28. Couch FJ, Sinilnikova O, Vierkant RA et al.: AURKA F31I polymorphism and breast cancer risk in BRCA1 and BRCA2 mutation carriers: a consortium of investigators of modifiers of BRCA1/2 study. Cancer Epidemiol Biomarkers Prev 2007 Jul; 16(7): 1416-1421.
29. Antoniou AC, Sinilnikova OM, Simard J et al.: RAD51 135G→C modifies breast cancer risk among BRCA2 mutation carriers: results from a combined analysis of 19 studies. Am J Hum Genet 2007 Dec; 81(6): 1186-1200. Epub 2007 Oct 16.
30. Marcus JN, Watson P, Page DL et al.: Hereditary breast cancer: pathobiology, prognosis, and BRCA1 and BRCA2 gene linkage. Cancer 1996; 77: 697-709.
31. Martin AM, Blackwood MA, Antin-Ozerkis D et al.: Germline mutations in BRCA1 and BRCA2 in breast-ovarian families from a breast cancer risk evaluation clinic. J Clin Oncol 2001; 19: 2247-2253.
32. Li FP, Fraumeni JF Jr: Soft-tissue sarcomas, breast cancer and other neoplasms. A familial syndrome? Ann Intern Med 1969; 71: 747-752.
33. Menkiszak J, Jakubowska A, Gronwald J et al.: Hereditary ovarian cancer: summary of 5 years of experience. Ginekol Pol 1998; 69: 283-287.
34. Loman N, Johannsson O, Bendahl P et al.: Prognosis and clinical presentation of BRCA2-associated breast cancer. Eur J Cancer 2000; 36: 1365-1373.
35. Lubinski J, Górski B, Huzarski T et al.: BRCA1-positive breast cancers in young women from Poland. Breast Cancer Res Treat 2006; 99: 71-76.
36. Lakhani SR: The pathology of familial breast cancer: Morphological aspects. Breast Cancer Res 1999; 1: 31-35.
37. Górski B, Byrski T, Huzarski T et al.: Founder mutations in the BRCA1 gene in Polish families with breast-ovarian cancer. Am J Hum Genet 2000; 66: 1963-1968.
38. Loman N, Johannsson O, Bendahl PO et al.: Steroid receptors in hereditary breast carcinomas associated with BRCA1 or BRCA2 mutations or unknown susceptibility genes. Cancer 1998; 83: 310-319.
39. Wooster R, Neuhausen SL, Mangion J et al.: Localisation of a breast cancer susceptibility gene BRCA2, to chromosome 13q12-13. Science 1994; 265: 2088-2090.
40. Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 in a population based series of breast cancer cases. Br J Cancer 2000; 83: 1301-1308.
41. Ford D, Easton DF, Stratton M et al.: Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. The Breast Cancer Linkage Consortium. Am J Hum Genet 1998; 62: 676-689.
42. Hopper JL, Southey MC, Dite GS et al.: Australian Breast Cancer Family Study. Population-based estimate of the average age specific cumulative risk of breast cancer for a defined set of protein-truncating mutations in BRCA1 and BRCA2. Cancer Epidemiol Biomarkers Prev 1999; 8: 741-747.
43. Warner E, Foulkes W, Goodwin P et al.: Prevalence and penetrance of BRCA1 and BRCA2 gene mutations in unselected Ashkenazi Jewish women with breast cancer. J Natl Cancer Inst 1999; 91: 1241-1247.
44. Jakubowska A, Nej K, Huzarski T et al.: BRCA2 gene mutation in families with aggregations of breast and stomach cancers. Br J Cancer 2002; 87: 888-891.
45. Jakubowska A, Scott R, Menkiszak J et al.: A high frequency of BRCA2 gene mutations in Polish families with ovarian and stomach cancer. Eur J Hum Genet 2003; 11: 955-958.
46. Kwiatkowska E, Teresiak M, Lamperska KM et al.: BRCA2 germline mutations in male breast cancer patients in the Polish population. Hum Mutat 2001; 17: 73.
47. Boyd J, Sonoda Y, Federici MG et al.: Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA 2000; 283: 2260-2265.
48. Malkin D, Li FP, Strong LC et al.: Germline p53 mutations in a familiar syndrome of breast cancer, sarcomas and other neoplasms. Science 1990; 250: 1233-1238.
49. Nelen MR, Padberg GW, Peeters EA et al.: Localization of the gene for Cowden disease to chromosome 10q22-23. Nat Genet 1996; 13: 114-116.
50. Hanssen AM, Fryns JP: Cowden syndrome. J Med Genet 1995; 32: 117-119.
51. Risinger JI, Barrett JC, Watson P et al.: Molecular genetic evidence of the occurrence of breast cancer as on integral tumor in patients with the hereditary nonpolyposis colorectal carcinoma syndrome. Cancer 1996; 77: 1836-1843.
52. Spigelman AD, Murday V, Phillips RK: Cancer and Peutz-Jeghers syndrome. Gut 1989; 30: 1588-1590.
53. Cohen MM Jr: A comprehensive and critical assessment of overgrowth and overgrowth syndromes. Adv Hum Genet 1989; 18: 181-303, 373-376.
54. Shiloh Y: Ataxia telangiectasia: closer to unraveling the mystery. Eur J Hum Genet 1995; 3: 116-138.
55. Swift M, Reitnauer PJ, Morrell D et al.: Breast and other cancers in families with ataxiatelangiectasia. N Engl J Med 1987; 316: 1289-1294.
56. Lynch HT, Kaplan AR, Lynch JF: Klinefelter syndrome and cancer. A family study. JAMA 1974; 229: 809-811.
57. Wooster R, Mangion J, Eeles R et al.: A germline mutation in the androgen receptor in two brothers with breast cancer and Reifenstein syndrome. Nat Genet 1992; 2: 132-134.
58. Serrano-Fernández P, Debniak T, Górski B et al.: Synergistic interaction of variants in CHEK2 and BRCA2 on breast cancer risk. Breast Cancer Res Treat 2009 Sep; 117(1): 161-165. doi: 10.1007/s10549-008-0249-1.
59. Gronwald J, Cybulski C, Piesiak W et al.: Cancer risks in first-degree relatives of CHEK2 mutation carriers: effects of mutation type and cancer site in proband. Br J Cancer 2009 May 5; 100(9): 1508-1512.
60. Cybulski C, Wokołorczyk D, Jakubowska A et al.: Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol 2011 Oct 1; 29(28): 3747-3752.
61. Narod SA, Risch H, Moslehi R et al.: Oral contraceptives and the risk of hereditary ovarian cancer. Hereditary Ovarian Cancer Clinical Study Group. N Engl J Med 1998; 339: 424-428.
62. Kotsopoulos J, Lubinski J, Moller P et al.: Timing of oral contraceptive use and the risk of breast cancer in BRCA1 mutation carriers. Breast Cancer Res Treat 2014 Feb; 143(3): 579-586.
63. McLaughlin JR, Risch HA, Lubinski J et al.: Hereditary Ovarian Cancer Clinical Study Group. Reproductive risk factors for ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet Oncol 2007; 8: 26-34.
64. Narod SA, Dubè MP, Klijn J et al.: Oral contraceptives and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst 2002; 94: 1773-1779.
65. Grabrick DM, Hartmann LC, Cerhan JR et al.: Risk of breast cancer with oral contraceptive use in women with a family history of breast cancer. JAMA 2000; 284: 1791-1798.
66. Armstrong K, Schwartz JS, Randall T et al.: Hormon replacement therapy and life expectancy after prophylactic oophorectomy in women with BRCA1/2 mutations: a decision analysis. J Clin Oncol 2004; 22: 1045-1054.
67. Rebbeck TR, Lynch HT, Neuhausen SL et al.: Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 2002; 346: 1616-1622.
68. Rebbeck TR, Friebel T, Wagner T et al.: Effect of Short-Term Hormone Replacement Therapy on Breast Cancer Risk Reduction After Bilateral Prophylactic Oophorectomy in BRCA1 and BRCA2 Mutation Carriers: The PROSE Study Group. J Clin Oncol 2005; 23: 7804-7810.
69. Narod SA: Hormonal prevention of hereditary breast cancer. Ann N Y Acad Sci 2001; 952: 36-43.
70. Gronwald J, Byrski T, Huzarski T et al.: Influence of selected lifestyle factors on breast and ovarian cancer risk in BRCA1 mutation carriers from Poland. Breast Cancer Res Treat 2006; 95: 105-109.
71. Kotsopoulos J, Lubinski J, Lynch HT et al.: Age at first birth and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat 2007; 105: 221-228.
72. Gronwald J, Tung N, Foulkes WD et al.: Hereditary Breast Cancer Clinical Study Group. Tamoxifen and contralateral breast cancer in BRCA1 and BRCA2 carriers: an update. Int J Cancer 2006; 118: 2281-2284.
73. Narod SA, Brunet JS, Ghadirian P et al.: Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: a case-control study. Hereditary Breast Cancer Clinical Study Group. Lancet 2000; 356: 1876-1881.
74. Gronwald J, Robidoux A, Kim-Sing C et al.: Duration of tamoxifen use and the risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat 2014 Jul; 146(2): 421-427.
75. Kotsopoulos J, Sukiennicki G, Muszyńska M et al.: Plasma micronutrients, trace elements, and breast cancer in BRCA1 mutation carriers: an exploratory study. Cancer Causes Control 2012 Jul; 23(7): 1065-1074.
76. Kotsopoulos J, Lubinski J, Lynch HT et al.: Oophorectomy after menopause and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Cancer Epidemiol Biomarkers Prev 2012 Jul; 21(7): 1089-1096.
77. Finch AP, Lubinski J, Møller P et al.: Impact of oophorectomy on cancer incidence and mortality in women with a BRCA1 or BRCA2 mutation. J Clin Oncol 2014 May 20; 32(15): 1547-1553.
78. Menkiszak J, Rzepka-Górska I, Górski B et al.: Attitudes toward preventive oophorectomy among BRCA1 mutation carriers in Poland. Eur J Gynaecol Oncol 2004; 25: 93-95.
79. Zeigler LD, Kroll SS: Primary breast cancer after prophylactic mastectomy. Am J Clin Oncol 1991; 14: 451-454.
80. Temple WJ, Lindsay RL, Magi E et al.: Technical considerations for prophylactic mastectomy in patients at high risk for breast cancer. Am J Surg 1991; 161: 413-415.
81. Warner E, Plewes DB, Shumak RS et al.: Comparison of breast magnetic resonance imaging, mammography, and ultrasound for surveillance of women at high risk for hereditary breast cancer. J Clin Oncol 2001; 19: 3524-3531.
82. Huzarski T, Byrski T, Gronwald J et al.: Ten-year survival in patients with BRCA1-negative and BRCA1-positive breast cancer. J Clin Oncol 2013 Sep 10; 31(26): 3191-3196.
83. Byrski T, Huzarski T, Dent R et al.: Response to neo-adjuvant chemotherapy in women with BRCA1-positive breast cancers. Breast Cancer Res Treat 2008; 108: 289-296.
84. Byrski T, Gronwald J, Huzarski T et al.: Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol 2010 Jan 20; 28(3): 375-379. Epub 2009 Dec 14.
85. Byrski T, Huzarski T, Dent R et al.: Pathologic complete response to neoadjuvant cisplatin in BRCA1-positive breast cancer patients. Breast Cancer Res Treat 2014 Sep; 147(2): 401-405.
86. Byrski T, Dent R, Blecharz P et al.: Results of a phase II open-label, non-randomized trial of cisplatin chemotherapy in patients with BRCA1-positive metastatic breast cancer. Breast Cancer Res 2012; 20; 14(4): R110.
87. Narod SA, Metcalfe K, Lynch HT et al.: Should all BRCA1 mutation carriers with stage I breast cancer receive chemotherapy? Breast Cancer Res Treat. 2013 Feb; 138(1): 273-279.
88. Lubinski J, Korzeń M, Górski B et al.: Genetic contribution to all cancers: the first demonstration using the model of breast cancers from Poland stratified by age at diagnosis and tumour pathology. Breast Cancer Res Treat 2009 Mar; 114(1): 121-126. Epub 2008 Apr 15.
89. Cybulski C, Górski B, Huzarski T et al.: CHEK2-positive breast cancers in young Polish women. Clin Cancer Res 2006; 12: 4832-4835.
90. Cybulski C, Wokołorczyk D, Huzarski T et al.: A deletion in CHEK2 of 5395 bp predisposes to breast cancer in Poland. Breast Cancer Res Treat 2007; 102: 119-122.
91. Huzarski T, Cybulski C, Domagała W et al.: Pathology of breast cancer in women with constitutional CHEK2 mutations. Breast Cancer Res Treat 2005; 90: 187-189.
92. Steffen J, Nowakowska D, Niwińska A et al.: Germline mutations 657del5 of the NBS1 gene contribute significantly to the incidence of breast cancer in Central Poland. Int J Cancer 2006; 119: 472-475.
93. Górski B, Debniak T, Masojć B et al.: Germline 657del5 mutation in the NBS1 gene in breast cancer patients. Int J Cancer 2003; 106: 379-381.
94. Huzarski T, Lener M, Domagala W et al.: The 3020insC allele of NOD2 predisposes to early onset breast cancer. Breast Cancer Res Treat 2005; 89: 91-93.
95. Menkiszak J, Lener M, Domagała W et al.: Clinical features of familial ovarian cancer lacking mutations in BRCA1 or BRCA2. Eur J Gynaecol Oncol 2004; 25: 99-100.
96. Hart WR: Mucinous tumors of the ovary: A review. Int J Gynecol Pathol 2005; 24: 4-25.
97. Shih I-M, Kurman RJ: Ovarian tumorigenesis: a proposed model based on morphological and molecular genetic analysis. Am J Pathol 2004; 164: 1511-1518.
98. Szymańska A: Identyfikacja genów związanych z predyspozycji do gruczolako-torbielaków śluzowych jajnika. Doctor’s thesis. Pomeranian Medical University, Szczecin 2006.
99. Szymańska-Pasternak J: Identyfikacja genów związanych z predyspozycją do gruczolako-torbielaków surowiczych jajnika. Doctor’s thesis. Pomeranian Medical University, Szczecin 2007.