© Borgis - Postępy Nauk Medycznych 7/2014, s. 479-486
*Andrzej Beręsewicz
Hypokalemia, aktywacja neurohormonalna i ryzyko groźnych arytmii
Hypokalemia, neurohormonal activation and arrhythmogenesis
Zakład Fizjologii Klinicznej, Centrum Medyczne Kształcenia Podyplomowego, Warszawa
Kierownik Zakładu: prof. dr hab. med. Andrzej Beręsewicz
Streszczenie
Hypokalemia, definiowana jako poziom K+ w surowicy poniżej 3,5 mM, jest częstym zaburzeniem elektrolitowym u pacjentów kardiologicznych. Hypokalemia jest czynnikiem ryzyka groźnych arytmii i nagłego zgonu sercowego a w badaniach potencjałów komórkowych wykazano, że skutkuje zmianami czynności elektrycznej serca, które powszechnie uznawane są za arytmogenne. Głównymi strażnikami ogólnoustrojowej homeostazy potasowej oraz stałości poziomu jonów K+ we krwi są: aldosteron – stymulujący nerkowe wydalanie K+, oraz insulina i katecholaminy – aktywujące błonową ATP-azę sodowo potasową, dokomórkowy transport K+ i wewnątrzkomórkową akumulację potasu. Patologie a także terapie, którym towarzyszą zmiany poziomów i/lub aktywności tych trzech substancji czynnych mogą skutkować zaburzeniami homeostazy K+ a następnie groźnymi arytmiami. Obok zaburzeń stricte endokrynologicznych, dotyczy to różnych chorób układu sercowo-naczyniowego. Przyczyną hypokalemii jest najczęściej leczenie lekami moczopędnymi i agonistami receptorów adrenergicznych b2 i/lub, częsta w chorobach układu sercowo-naczyniowego, aktywacja układu współczulnego i układu renina-angiotensyna-aldosteron. Obecny artykuł opisuje zaburzenia homeostazy potasowej towarzyszące chorobom kardiologicznym oraz potencjalne mechanizmy elektrofizjologiczne arytmii towarzyszących hypokalemii.
Summary
Main regulators of potassium body homeostasis and, in particular, of serum K+ concentration, include: aldosteron, which stimulates renal K+ excretion, and catecholamines and insulin, which, via the membrane Na+/K+ ATP-ase activation, mediate intracellular K+ transport and cellular potassium accumulation. Various pathologies and therapies that are associated with altered level and/or activity of these three mediators are known to result in disturbed potassium homeostasis. This is true for genuine endocrinologic disorders as well as for cardiovascular disease. Hypokalemia, defined as serum K+ concentration < 3.5 mM, is a common biochemical finding in cardiac patients recognized as an independent risk-factor of severe cardiac arrhythmias and sudden cardiac death. Most often mechanisms of hypokalemia include diuretic therapy, therapy with agonists of adrenergic β2 receptors, and frequent in cardiovascular disease, sympathetic activation and activation of the rennin-angiotensin-aldosterone system. Present article reviews the mechanisms of potassium body homeostasis as well as potential electrophysiological mechanisms underlying hypokalemia-induced arrhythmogenesis.

WSTĘP
Nagły zgon sercowy (NZS), definiowany jako „naturalny zgon spowodowany niespodziewanym zatrzymaniem krążenia i występujący w ciągu 1 godziny od pojawienia się objawów choroby lub w czasie snu”, jest problemem o skali epidemicznej (~10% rocznej śmiertelności, 1 zgon/1000 osobników populacji generalnej rocznie, w Polsce ~50 tys. NZS rocznie) (1-3). Przyczyną NZS są głównie groźne zaburzenia rytmu, takie jak częstoskurcz komorowy i migotanie komór (2, 4-7). Dlatego identyfikacja mechanizmów powstawania i opracowanie metod prewencji tych arytmii jest ważnym wyzwaniem dla współczesnej kardiologii.
NZS może być traktowany jako „wypadek” elektrofizjologiczny, którego zaistnienie wymaga „zbiegu następujących trzech okoliczności”: (a) obecności substratu – w postaci organicznej choroby serca skutkującej zmianami anatomicznymi, takimi jak blaszki miażdżycowe, blizny, zwłóknienie miokardium, przebudowa elektryczna miokardium itp., (b) obecności czynnika wyzwalającego – w postaci zmian środowiska życiowego miocytów i/lub komórek układu His-Purkinje (niedokrwienie/reperfuzja, zaburzenia elektrolitowe z hypokalemią na czele, aktywacja współczulna sama i/lub skutkująca hypokalemią itp.) wywołujących potencjalnie arytmogenne zaburzenia czynności elektrycznej komórek sercowych, oraz (c) specjalnej konstelacji zmian elektrofizjologicznych potrzebnych do wystąpienia mechanizmu pobudzenia krążącego (re-entry) stanowiącego podstawę > 90% groźnych arytmii takich jak częstoskurcz komorowy i migotanie komór (ryc. 1). Za słusznością tego trójskładnikowego modelu NZS przemawiają dwa fakty. Po pierwsze, powszechnie występujące komorowe pobudzenia dodatkowe (PVC) i nieutrwalony częstoskurcz komorowy (NSVT) pozostają bez wpływu na ryzyko występowania NZS w populacji osób zdrowych, a są predykatorem występowania NZS wśród osób z organicznymi chorobami serca (2). Podobnie Farmingham Heart Study wykazało, że hypokalemia nie ma wpływu na rokowanie u osób zdrowych (8). Jest natomiast niezależnym czynnikiem ryzyka występowania arytmii u pacjentów z chorobą sercowo-naczyniową (9, 10). Tematem obecnego przeglądu literatury jest hypokalemia, o której wiadomo, że jest bardzo częstym zaburzeniem elektrolitowym w chorobach sercowo-naczyniowych związanym z zaburzeniami neurohormonalnymi towarzyszącymi tym chorobom oraz że jest czynnikiem ryzyka groźnych arytmii, ale również czynnikiem skutkującym proarytmicznymi komórkowymi zmianami elektrofizjologicznymi.
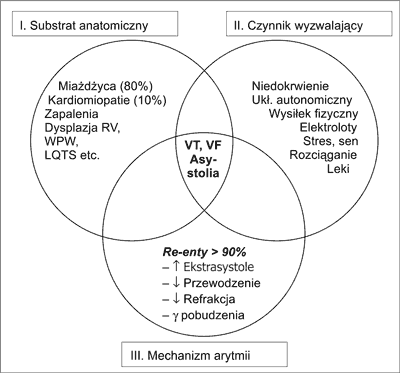
Ryc. 1. Trójskładnikowy model mechanizmu nagłego zgonu sercowego (NZS). Warunkiem wystąpienia groźnych arytmii, takich jak częstoskurcz komorowy (VT) i migotanie komór (VF), będących najczęstszymi przyczynami NZS, jest koincydencja organicznej choroby serca (substrat anatomiczny), zaburzeń środowiska komórek sercowych (czynnik wyzwalający) oraz zmian elektrofizjologicznych warunkujących wystąpienie mechanizmu pobudzenia krążącego (re-entry).
HYPOKALEMIA – SKALA PROBLEMU
Hypokalemia, definiowana jako poziom K+ w surowicy poniżej 3,5 mM, jest częstym zaburzeniem elektrolitowym. Dla przykładu, jej obecność obserwowano u ~20% pacjentów przebywających aktualnie w szpitalu (11), u 10-40% osób leczonych lekami moczopędnymi niezatrzymującymi potasu, zwłaszcza lekami tiazydowymi (9) oraz u 7-17% pacjentów z różnymi postaciami choroby sercowo-naczyniowej (tab. 1) (9).
Tabela 1. Występowanie hypokalemii w chorobie sercowo-naczyniowej.
| Rozpoznanie | Liczba pacjentów | Pacjenci z hypokalemią | Referencja |
| Nadciśnienie tętnicze | 2102
3156 | 151 (7%)
225 (7,1%)
(leczeni diuretyk. [12,9%]) | (12)
(13) |
| Niewydolność serca | 3313
6845 | 431 (13%)
1189 (17%)* | (14)
(15) |
| Ostry zawał serca | 1074
1011
517 | 122 (11%)
131 (13%)
41 (8%) | (16)
(17)
(18) |
| Resuscytacja po zatrzymaniu krążenia poza szpitalem | 132 | 54 (41%) | (19) |
*W oznaczonym badaniu hypokalemię definiowano jako K+ w surowicy < 4,0 mM, a w pozostałych jako K+ < 3,5 mM.
Najczęstsze przyczyny hypokalemii w kardiologii to: (a) leczenie diuretykami niezatrzymującymi potasu, (b) aktywacja endogennego układu renina-angiotensyna-aldosteron (RAA) oraz (c) aktywacja układu współczulnego (9-13). W rzeczywistości czynniki te najczęściej współistnieją u pacjentów kardiologicznych, więc ich efekty mogą się nawzajem wzmacniać (tab. 2).
Tabela 2. Najczęstsze przyczyny hypokalemii u pacjentów kardiologicznych.
| I. Ustrojowa utrata K+ |
– Diuretyki niezatrzymujące K+ i wtórny hiperaldosteronizm
– Przewlekła aktywacja układu RAA (aldosteron)
– Wymioty, biegunka, nadmierne pocenie i wtórny hiperaldosteronizm
– Mała podaż K+ i Mg2+ w diecie, niedożywienie
– Alkaloza |
| II. Aktywacja pompy Na+/K+ i przemieszczenie K+ do mięśni |
– Aktywacja współczulna (zawał, niewydolność serca, uraz, sepsa, pheochromocytoma, nadciśnienie, bezdech nocny, diuretyki)
– Katecholaminy
– Agoniści β2-receptorów (leczenie astmy, doping w sporcie)
– Metyloksantyny (kofeina, teofilina, coca-cola)
– Hyperinsulinemia |
USTROJOWA HOMEOSTAZ JONÓW K+
Potas bierze udział w regulacji objętości komórek, regulacji pH komórkowego, a także regulacji aktywności różnych enzymów, w tym ATP-azy sodowo-potasowej (pompy Na+/K+). Aktywność pompy Na+/K+ i duży przezbłonowy gradient K+ (wnętrze ~140 mM, zewnętrze 3,5-5,0 mM) decydują o potencjale spoczynkowym komórek, a w przypadku tkanek pobudliwych (tkanka nerwowa, mięśnie, serce) także o ich pobudliwości i przewodzeniu impulsów elektrycznych. Każde zaburzenie przezbłonowego gradientu K+, czy to poprzez zmianę zewnątrzkomórkowego stężenia K+, czy komórkowej zawartości K+ może skutkować zaburzeniami czynności tych tkanek. Dodatkowo, na drodze nieznanego mechanizmu hypokalemia jest czynnikiem indukującym naczyniowy stres oksydacyjny, a wzrost poziomu K+ we krwi zmniejsza naczyniową produkcję wolnych rodników (14).
Sód jest jonem głównie pozakomórkowym, a K+ wewnątrzkomórkowym. Około 98% ustrojowych zasobów K+ (~3600 mmoli) znajduje się w komórkach, w tym, ze względu na ich dużą masę, głównie w mięśniach szkieletowych (2600 mmoli), a jedynie 2% w płynie zewnątrzkomórkowym (~65 mmoli) (ryc. 2) (12-14). Fakt, że pula zewnątrzkomórkowego K+ jest bardzo mała (~2% całości), sprawia, że pomiary zawartości K+ we krwi bardzo słabo informują o komórkowej zawartości K+. Generalnie przyjmuje się, że obniżenie stężenia potasu w surowicy o kolejne 0,3 mM oznacza zubożenie ustrojowych zasobów K+ o kolejne ~100 mmoli/l. Stąd stężenie K+ w surowicy ~3 mM oznaczałoby deficyt ustrojowy K+ wynoszący ~200 mmoli/l (12).
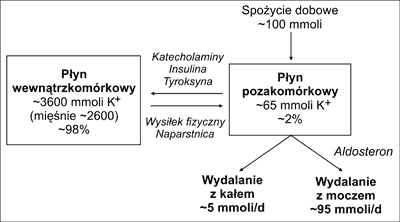
Ryc. 2. Ustrojowa homeostaza K+. W płynie wewnątrz- i zewnątrzkomórkowym znajduje się odpowiednio ~98% i ~2% ustrojowego K+. Poziom zewnątrzkomórkowego K+ jest wypadkową: (a) wchłaniania z przewodu pokarmowego K+ dostarczanego z dietą, (b) eliminacji K+ z moczem i w mniejszym stopniu ze stolcem oraz (c) przemieszczania się K+ między przestrzenią zewnątrz- i wewnątrzkomórkową. Głównymi regulatorami homeostazy K+ są: aldosteron, insulina i katecholaminy.
Pula zewnątrzkomórkowego K+ podlega stałej intensywnej wymianie i jest ściśle regulowana. W życiu codziennym głównymi czynnikami zakłócającymi homeostazę K+ są dieta (dzienna pokarmowa podaż K+ wynosi ~100 mmoli/l, a zawartość K+ w płynie zewnątrzkomórkowym to jedynie 65 mmoli/l) (ryc. 2) oraz wysiłek fizyczny. Komórki mięśni szkieletowych charakteryzują się dużą ekspresją kanałów potasowych, przez które K+ wypływa w czasie skurczu, a także dużą ekspresją pompy Na+/K+, która transportuje K+ z powrotem do komórek. Ze względu na dysproporcje w wielkości pul K+, niewielkie zmiany w puli wewnątrzkomórkowego K+ skutkują ogromnymi zmianami stężenia K+ w przestrzeni zewnątrzkomórkowej. Dla przykładu, w czasie maksymalnego wysiłku mięśnie tracą ~40 mmoli/min K+, co oznacza, że stężenie K+ we krwi może się w tym czasie prawie podwoić. Z drugiej strony, obliczenia wskazują, że maksymalna aktywacja pompy Na+/K+ obecnej w mięśniach całego ciała mogłaby teoretycznie skutkować dokomórkowym transportem ~130 mmoli/min K+, co w ciągu ~30 sekund zredukowałoby poziom K+ we krwi do zera (13).
Głównymi strażnikami stałości poziomu K+ we krwi i ogólnie homeostazy potasowej są: aldosteron – stymulujący nerkowe wydalanie K+, oraz insulina i katecholaminy – aktywujące dokomórkowy transport K+ i jego wewnątrzkomórkową akumulację (ryc. 2) (11-13). Patologie, a także terapie, którym towarzyszą zmiany poziomów i/lub aktywności tych trzech substancji czynnych, mogą skutkować zaburzeniami homeostazy K+. Dotyczy to różnych chorób endokrynologicznych (12), ale także kardiologicznych (tab. 2).
Na poposiłkową regulację homeostazy K+ składają się trzy elementy: (a) K+ w pokarmie na drodze sygnalizacji przewód pokarmowy-nerka skutkuje zwiększonym wydalaniem K+ przez nerki. Mechanizm tej sygnalizacji jest niejasny, ale wyprzedza ona w czasie poposiłkowy wzrost poziomu K+ i aldosteronu we krwi (15); (b) poposiłkowy wzrost poziomu K+ we krwi stymuluje produkcję aldosteronu, który aktywuje nerkowe wydalanie K+, oraz (c) poposiłkowy wzrost poziomu K+ we krwi stymuluje produkcję insuliny, która aktywuje komórkową sekwestrację K+. Istnieje sprzężenie zwrotne między poziom K+ we krwi i wydzielaniem aldosteronu i insuliny, hyperkalemia wydzielanie to aktywuje, a hypokalemia je hamuje (11-13).
Aldosteron uczestniczy w bieżącej regulacji homeostazy K+, ale w różnych stanach patologicznych może być przyczyną dysregulacji tej homeostazy. Dla przykładu, główną przyczyną utraty K+ i hypokalemii u pacjentów leczonych diuretykami tiazydowymi i pętlowymi jest, wtórne do podiuretykowej utraty Na+ i hipowolemii, zwiększone uwalnianie reniny, aktywacja układu RAA i stymulowana przez aldosteron nerkowa utrata K+ (16). Zgodnie z tym mechanizmem, inhibitory receptora aldosteronowego skutecznie redukują częstość podiuretykowej hypokalemii, a także występowanie arytmii komorowych i NZS u pacjentów leczonych diuretykami. Natomiast zwykła suplementacja K+ nie miała takiego efektu (9, 16). Podobnie, spowodowana hipowolemią aktywacja układu RAA i wtórny hiperaldosteronizm jest ważną częścią mechanizmu hypokalemii towarzyszącej intensywnym wymiotom i biegunkom (12). Obok hipowolemii (hipotonii), innymi ważnymi czynnikami pobudzającymi wydzielanie reniny i aktywność całego układu RAA są hiponatremia i aktywacja współczulna (poprzez aktywację receptorów β-adrenergicznych) (17, 20). Z drugiej strony angiotensyna II i aldosteron, działając na różnych poziomach, skutkują zwiększoną aktywacją współczulną (17-19). Na udział układów RAA i układu współczulnego w dysregulacji homeostazy K+ u pacjentów kardiologicznych wskazuje fakt, że leki blokujące aktywność układu RAA oraz β-blokery zwiększają stężenie K+ w surowicy (10, 12, 13) lub zapobiegają hypokalemii, ostremu niedokrwieniu i zawałowi serca (9).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Chugh SS, Jui J, Gunson K et al.: Current burden of sudden cardiac death: multiple source surveillance versus retrospective death certificate-based review in a large U.S. community. J Am Coll Cardiol 2004; 44: 1268-1275.
2. Zipes DP, Camm AJ, Borggrefe M et al.: ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: a report of the American College of Cardiology/American Heart Association Task Force and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Develop Guidelines for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death). J Am Coll Cardiol 2006; 48: e247-e346.
3. Fishman GI, Chugh SS, DiMarco JP et al.: Sudden cardiac death prediction and prevention: report from a National Heart, Lung, and Blood Institute and Heart Rhythm Society Workshop. Circulation 2010; 122: 2335-2348.
4. Bayes dL, Coumel P, Leclercq JF: Ambulatory sudden cardiac death: mechanisms of production of fatal arrhythmia on the basis of data from 157 cases. Am Heart J 1989; 117(1): 151-159.
5. Olshausen KV, Witt T, Pop T et al.: Sudden cardiac death while wearing a Holter monitor. Am J Cardiol 1991; 67: 381-386.
6. Myerburg RJ, Spooner PM: Opportunities for sudden death prevention: directions for new clinical and basic research. Cardiovasc Res 2001; 50: 177-185.
7. John RM, Tedrow UB, Koplan BA et al.: Ventricular arrhythmias and sudden cardiac death. Lancet 2012; 380: 1520-1529.
8. Walsh CR, Larson MG, Leip EP et al.: Serum potassium and risk of cardiovascular disease: the Framingham heart study. Arch Intern Med 2002; 162: 1007-1012.
9. Osadchii OE: Mechanisms of hypokalemia-induced ventricular arrhythmogenicity. Fundam Clin Pharmacol 2010; 24: 547-559.
10. Kjeldsen K: Hypokalemia and sudden cardiac death. Exp Clin Cardiol 2010; 15: e96-e99.
11. Gennari FJ: Hypokalemia. N Engl J Med 1998; 339: 451-458.
12. Unwin RJ, Luft FC, Shirley DG: Pathophysiology and management of hypokalemia: a clinical perspective. Nat Rev Nephrol 2011; 7: 75-84.
13. Clausen T: Hormonal and pharmacological modification of plasma potassium homeostasis. Fundam Clin Pharmacol 2010; 24: 595-605.
14. Youn JH, McDonough AA: Recent advances in understanding integrative control of potassium homeostasis. Annu Rev Physiol 2009; 71: 381-401.
15. Youn JH: Gut sensing of potassium intake and its role in potassium homeostasis. Semin Nephrol 2013; 33: 248-256.
16. Laragh JH, Sealey JE: K(+) depletion and the progression of hypertensive disease or heart failure. The pathogenic role of diuretic-induced aldosterone secretion. Hypertension 2001; 37: 806-810.
17. Weir MR, Dzau VJ: The renin-angiotensin-aldosterone system: a specific target for hypertension management. Am J Hypertens 1999; 12: 205S-213S.
18. Zucker IH: Novel mechanisms of sympathetic regulation in chronic heart failure. Hypertension 2006; 48: 1005-1011.
19. Gao L, Wang W, Li YL et al.: Sympathoexcitation by central ANG II: roles for AT1 receptor upregulation and NAD(P)H oxidase in RVLM. Am J Physiol 2005; 288: H2271-H2279.
20. Castrop H, Hocherl K, Kurtz A et al.: Physiology of kidney renin. Physiol Rev 2010; 90: 607-673.
21. DeFronzo RA, Felig P, Ferrannini E, Wahren J: Effect of graded doses of insulin on splanchnic and peripheral potassium metabolism in man. Am J Physiol 1980; 238: E421-E427.
22. DeFronzo RA: Obesity is associated with impaired insulin-mediated potassium uptake. Metabolism 1988; 37: 105-108.
23. Arslanian S, Austin A: Impaired insulin mediated potassium uptake in adolescents with IDDM. Biochem Med Metab Biol 1991; 46: 364-372.
24. Medbo JI, Sejersted OM: Plasma potassium changes with high intensity exercise. J Physiol 1990; 421: 105-122.
25. Terkildsen JR, Crampin EJ, Smith NP: The balance between inactivation and activation of the Na+-K+ pump underlies the triphasic accumulation of extracellular K+ during myocardial ischemia. Am J Physiol Heart Circ Physiol 2007; 293: H3036-H3045.
26. Wong CS, Pavord ID, Williams J et al.: Bronchodilator, cardiovascular, and hypokalaemic effects of fenoterol, salbutamol, and terbutaline in asthma. Lancet 1990; 336: 1396-1399.
27. Salpeter SR, Ormiston TM, Salpeter EE: Cardiovascular effects of beta-agonists in patients with asthma and COPD: a meta-analysis. Chest 2004; 125: 2309-2321.
28. Evans K, Reddan DN, Szczech LA: Nondialytic management of hyperkalemia and pulmonary edema among end-stage renal disease patients: an evaluation of the evidence. Semin Dial 2004; 17: 22-29.
29. Packer CD: Cola-induced hypokalaemia: a super-sized problem. Int J Clin Pract 2009; 63: 833-835.
30. Sharma R, Guber HA: Cola-induced hypokalemia-a case report and review of the literature. Endocr Pract 2013; 19: e21-e23.
31. Foo K, Sekhri N, Deaner A et al.: Effect of diabetes on serum potassium concentrations in acute coronary syndromes. Heart 2003; 89: 31-35.
32. Robinson RT, Harris ND, Ireland RH et al.: Mechanisms of abnormal cardiac repolarization during insulin-induced hypoglycemia. Diabetes 2003; 52: 1469-1474.
33. El-Sherif N, Turitto G: Electrolyte disorders and arrhythmogenesis. Cardiol J 2011; 18: 233-245.
34. Paltiel O, Salakhov E, Ronen I et al.: Management of severe hypokalemia in hospitalized patients: a study of quality of care based on computerized databases. Arch Intern Med 2001; 161: 1089-1095.
35. Cohen JD, Neaton JD, Prineas RJ, Daniels KA: Diuretics, serum potassium and ventricular arrhythmias in the Multiple Risk Factor Intervention Trial. Am J Cardiol 1987; 60: 548-554.
36. Siscovick DS, Raghunathan TE, Psaty BM et al.: Diuretic therapy for hypertension and the risk of primary cardiac arrest. N Engl J Med 1994; 330: 1852-1857.
37. Cohen HW, Madhavan S, Alderman MH: High and low serum potassium associated with cardiovascular events in diuretic-treated patients. J Hypertens 2001; 19: 1315-1323.
38. Nordrehaug JE: Malignant arrhythmia in relation to serum potassium in acute myocardial infarction. Am J Cardiol 1985; 56: 20D-23D.
39. Friedensohn A, Faibel HE, Bairey O et al.: Malignant arrhythmias in relation to values of serum potassium in patients with acute myocardial infarction. Int J Cardiol 1991; 32: 331-338.
40. Madias JE, Shah B, Chintalapally G et al.: Admission serum potassium in patients with acute myocardial infarction: its correlates and value as a determinant of in-hospital outcome. Chest 2000; 118: 904-913.
41. Ahmed A, Zannad F, Love TE et al.: A propensity-matched study of the association of low serum potassium levels and mortality in chronic heart failure. Eur Heart J 2007; 28: 1334-1343.
42. Cooper HA, Dries DL, Davis CE et al.: Diuretics and risk of arrhythmic death in patients with left ventricular dysfunction. Circulation 1999; 100: 1311-1315.
43. Ekundayo OJ, Adamopoulos C, Ahmed MI et al.: Oral potassium supplement use and outcomes in chronic heart failure: a propensity-matched study. Int J Cardiol 2010; 141: 167-174.
44. Krijthe BP, Heeringa J, Kors JA et al.: Serum potassium levels and the risk of atrial fibrillation: the Rotterdam Study. Int J Cardiol 2013; 168: 5411-5415.
45. Wolk R, Kane KA, Cobbe SM, Hicks MN: Apex-to-base dispersion of refractoriness underlies the proarrhythmic effect of hypokalaemia/hypomagnesaemia in the rabbit heart. J Electrocardiol 2002; 35: 245-252.
46. Wolk R, Kane KA, Cobbe SM, Hicks MN: Regional electrophysiological effects of hypokalaemia, hypomagnesaemia and hyponatraemia in isolated rabbit hearts in normal and ischaemic conditions. Cardiovasc Res 1998; 40: 492-501.
47. Reiter MJ, Mann DE, Williams GR: Interaction of hypokalemia and ventricular dilatation in isolated rabbit hearts. Am J Physiol 1993; 265: H1544-H1550.
48. Dominguez G, Fozzard HA: Influence of extracellular K+ concentration on cable properties and excitability of sheep cardiac Purkinje fibers. Circ Res 1970; 26: 565-574.
49. Killeen MJ, Gurung IS, Thomas G et al.: Separation of early afterdepolarizations from arrhythmogenic substrate in the isolated perfused hypokalaemic murine heart through modifiers of calcium homeostasis. Acta Physiol (Oxf) 2007; 191: 43-58.
50. Sicouri S, Antzelevitch C: Electrophysiologic characteristics of M cells in the canine left ventricular free wall. J Cardiovasc Electrophysiol 1995; 6: 591-603.
51. Gilat E, Nordin CW, Aronson RS: The role of reduced potassium conductance in generating triggered activity in guinea-pig ventricular muscle. J Mol Cell Cardiol 1990; 22: 619-628.
52. Ohta M, Karagueuzian HS, Mandel WJ, Peter T: Acute and chronic effects of amiodarone on delayed afterdepolarization and triggered automaticity in rabbit ventricular myocardium. Am Heart J 1987; 113: 289-296.
53. Christe G: Effects of low (K+)o on the electrical activity of human cardiac ventricular and Purkinje cells. Cardiovasc Res 1983; 17: 243-250.
54. Poelzing S, Veeraraghavan R: Heterogeneous ventricular chamber response to hypokalemia and inward rectifier potassium channel blockade underlies bifurcated T wave in guinea pig. Am J Physiol Heart Circ Physiol 2007; 292: H3043-H3051.
55. Franse LV, Pahor M, Di Bari M et al.: Hypokalemia associated with diuretic use and cardiovascular events in the Systolic Hypertension in the Elderly Program. Hypertension 2000; 35: 1025-1030.
56. Alderman MH, Piller LB, Ford CE et al.: Clinical significance of incident hypokalemia and hyperkalemia in treated hypertensive patients in the antihypertensive and lipid-lowering treatment to prevent heart attack trial. Hypertension 2012; 59: 926-933.
57. Pitt B, Bakris G, Ruilope LM et al.: Serum potassium and clinical outcomes in the Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS). Circulation 2008; 118: 1643-1650.
58. Salerno DM, Asinger RW, Elsperger J et al.: Frequency of hypokalemia after successfully resuscitated out-of-hospital cardiac arrest compared with that in transmural acute myocardial infarction. Am J Cardiol 1987; 59: 84-88.
59. Osadchii OE, Bentzen BH, Olesen SP: Chamber-specific effects of hypokalaemia on ventricular arrhythmogenicity in isolated, perfused guinea-pig heart. Exp Physiol 2009; 94: 434-446.
60. Osadchii OE, Larsen AP, Olesen SP: Predictive value of electrical restitution in hypokalemia-induced ventricular arrhythmogenicity. Am J Physiol Heart Circ Physiol 2010; 298: H210-H220.
61. Sabir IN, Li LM, Grace AA, Huang CL: Restitution analysis of alternans and its relationship to arrhythmogenicity in hypokalaemic Langendorff-perfused murine hearts. Pflugers Arch 2008; 455: 653-666.
62. Katzung BG, Morgenstern JA: Effects of extracellular potassium on ventricular automaticity and evidence for a pacemaker current in mammalian ventricular myocardium. Circ Res 1977; 40: 105-111.
63. Osadchii OE, Olesen SP: Electrophysiological determinants of hypokalaemia-induced arrhythmogenicity in the guinea-pig heart. Acta Physiol (Oxf) 2009; 197: 273-287.