© Borgis - Postępy Nauk Medycznych 7/2014, s. 461-469
*Andrzej Beręsewicz
Pozasercowe mechanizmy objawów niewydolności serca. Udział układu współczulnego i mięśni szkieletowych
Extracardiac mechanisms of heart failure symptoms. Involvement of sympathetic nervous system and skeletal muscles
Zakład Fizjologii Klinicznej, Centrum Medyczne Kształcenia Podyplomowego, Warszawa
Kierownik Zakładu: prof. dr hab. med. Andrzej Beręsewicz
Streszczenie
Niewydolność serca jest chorobą wielonarządową, a nie wyłącznie hemodynamiczną. Bezpośrednim źródłem głównych objawów niewydolności serca takich jak męczliwość i duszność jest miopatia mięśni szkieletowych (w tym miopatia przepony i mieśni międzyżebrowych) wtórna do zwiększonej aktywacji współczulnej skutkującej przykurczem tętnic, co ogranicza wysiłkową perfuzję mięśni szkieletowych. W czasie wysiłku, układ krążenia utrzymuje względnie stałe ciśnienie tętnicze (ciśnienie perfuzyjne mózgu, serca, mięśni oddechowych i innych) poprzez ścisłe dostosowywanie mięśniowego oporu naczyniowego do wielkości rzutu minutowego. Redukcja wysiłkowego rzutu minutowego w niewydolności serca wymusza zwiększony neurogenny skurcz naczyń w pracujących mięśniach. Skutkuje to powtarzającymi się incydentami niedokrwienia/reperfuzji pracujących mięśni, ich miopatią a następnie zwiększoną aktywnością ergoreceptorów mięśniowych, które jeszcze bardziej aktywują układ współczulny. Trening fizyczny zmniejsza objawy niewydolności serca prawdopodobnie, dlatego, że przerywa to błędne koło a nie, dlatego że poprawia czynność serca. Trening, prawdopodobnie poprzez poprawę czynności śródbłonka naczyniowego, zmniejsza aktywację współczulną, co skutkuje poprawą ukrwienia i czynności mięśni szkieletowych i ich lepszą tolerancją wysiłku.
Summary
In chronic heart failure from systolic cardiac dysfunction (HF), the degree of exercise intolerance is not directly related to the degree of cardiac weakness, but rather to intrinsic alterations in skeletal musculature. This skeletal myopathy is likely mediated by increased activity of the sympathetic nervous system associating HF and by consequent muscle hypoperfusion and injury. During intensive exercise, muscular vascular conductance must be restrained by neural sympathetic vasoconstriction to match actual cardiac output to avoid hypotension. In HF patients, characterized by an impaired cardiac output reserve, this neurogenic restrain must be even stronger. This results in repeated incidences of ischemia/reperfusion of the exercising muscles, their myopathy, and in increased activation of ergoreceptors in the myopathic muscles further enhancing sympathetic outflow and perpetuating muscular alterations. Exercise training improves symptoms and exercise capacity in HF patients by stopping this vicious circle. It is hypothesized that exercise by increasing endothelial shear stress, upregulates endothelial nitric oxide generation that, in turn, acts to restrain sympathetic outflow.

MIĘŚNIOWY MECHANIZM NIETOLERANCJI WYSIŁKU W NIEWYDOLNOŚCI SERCA
Przewlekła niewydolność serca (NS) jest zespołem chorobowym, któremu towarzyszą zobiektywizowane cechy organicznej choroby serca oraz typowe objawy podmiotowe (ang. symptoms), takie jak zwiększona męczliwość mięśni szkieletowych (ang. fatigue) i/lub duszność wysiłkowa, które obniżają tolerancję wysiłku i pogarszają jakość życia pacjentów (tab. 1). U ~50% osób z NS dominują zaburzenia skurczu i, wobec tego, opróżniania komór, a u pozostałych ~50% – zaburzenia rozkurczu i wypełniania komór. Stąd rozróżnienie na tzw. NS skurczową (NS z obniżoną frakcją wyrzucania) i rozkurczową (NS z zachowaną frakcją wyrzucania).
Tabela 1. Klasyfikacja objawów podmiotowych (symptoms) niewydolności serca według New York Heart Association (NYHA).
| Klasa I | Bez ograniczenia zwykłej aktywności fizycznej. |
| Klasa II | Niewielkie ograniczenie aktywności fizycznej.
Komfort w spoczynku. Zwykła aktywność powoduje zmęczenie, kołatanie serca lub duszność. |
| Klasa III | Znaczne ograniczenie aktywności fizycznej.
Komfort w spoczynku. Aktywność mniejsza niż przeciętna powoduje zmęczenie, kołatanie serca lub duszność. |
| Klasa IV | Każda aktywność fizyczna powoduje dyskomfort.
Objawy obecne już w spoczynku. Każda aktywność fizyczna zwiększa dyskomfort. |
Geneza nietolerancji wysiłku w NS jest tradycyjnie tłumaczona w oparciu o „kardio-centryczny” model NS. Zakłada on, że z powodu niedomogi serca wysiłkowy wzrost rzutu minutowego jest upośledzony i dlatego perfuzja pracujących mięśni jest za mała w stosunku do potrzeb. Dochodzi do niedokrwienia i gromadzenia się w mięśniach kwasu mlekowego i innych metabolitów, co skutkuje reakcją odczuwaną jako zmęczenie mięśni. W przypadku skurczowej NS, rosnące zaleganie krwi w lewej komorze i wzrost jej ciśnienia napełniania (mechanizm częściowo kompensacyjny, zwiększający rekrutację mechanizmu Franka-Starlinga) mogą skutkować wzrostem ciśnienia w krążeniu płucnym, zmniejszoną podatnością płuc, upośledzeniem wymiany gazowej i ewentualnym przesiękiem do pęcherzyków płucnych, co ostatecznie skutkuje uczuciem duszności. Paradoks polega na tym, że objawy, będące rzekomo bezpośrednią konsekwencją niedomogi serca, maleją pod wpływem treningu fizycznego, stanowiącego dodatkowe obciążenie serca (1-3). Rekomendacja Europejskiego Towarzystwa Kardiologicznego na ten temat brzmi następująco: „Pacjentów z NS należy zachęcać do regularnych aerobowych ćwiczeń ruchowych celem poprawy ich wydolności fizycznej oraz redukcji objawów podmiotowych (symptoms)” (klasa rekomendacji IA) (4). Według aktualnej interpretacji tego paradoksu w NS w czasie wysiłku mięśnie ulegają niedokrwieniu nie dlatego, że pompa sercowa jest zbyt słaba, by tłoczyć krew, ale dlatego, że zbyt duży opór mięśniowy uniemożliwia odpowiednią do potrzeb perfuzję mięśni (model „mięśniocentryczny”). Przyczyna tkwi w uogólnionej miopatii mięśni szkieletowych, jaka często towarzyszy NS. Trening fizyczny działa głównie na mięśnie, a nie na serce, i prawdopodobnie dlatego łagodzi objawy NS (1, 3, 5-7).
WYSIŁKOWA REGULACJA KRĄŻENIA I ZASADA STAŁOŚCI CIŚNIENIA TĘTNICZEGO
Centralnej aktywacji pracy mięśniowej towarzyszy aktywacja układu współczulnego, która ulega dodatkowemu wzmocnieniu w wyniku odruchu z ergoreceptorów (8), co razem ma na celu utrzymanie homeostazy krążeniowo-oddechowej w czasie wysiłku. Pracy mięśni towarzyszy aktywacja mięśniowych receptorów wrażliwych na mechaniczne odkształcenia (mechanoreceptory aktywujące włókna dośrodkowe typu III) oraz na gromadzące się w mięśniach metabolity, takie jak H+, K+ i kwas mlekowy (metaboreceptory aktywujące włókna aferentne typu IV). Aktywacja tych tzw. ergoreceptorów, poprzez aktywację ośrodka współczulnego w rdzeniu przedłużonym, inicjuje odruch z ergoreceptorów, którego składowymi są (8):
1. Stymulacja ośrodka oddechowego i wzrost wentylacji płucnej.
2. Skurcz mięśni gładkich tętniczek oporowych w nerkach, krążeniu trzewnym i niepracujących mięśniach i spadek perfuzji w tych narządach, wtórny do aktywacji naczyniowych α1-receptorów adrenergicznych.
3. Analogiczny skurcz tętniczek oporowych w pracujących mięśniach, który jest jednak częściowo antagonizowany przez lokalnie działające czynniki, w tym głównie ATP (czynnościowa sympatykoliza) (9, 10). Rozkurcz zwykle przeważa nad skurczem, co skutkuje lokalnym wzrostem perfuzji pracujących mięśni (wysiłkowa hyperemia), a w dalszej konsekwencji wzrostem powrotu żylnego, aktywacją mechanizmu Franka-Starlinga i wzrostem rzutu minutowego (11, 12). Głównym regulatorem wysiłkowej hyperemii jest ATP, działający poprzez receptory purynergiczne 2Pγ. ATP jest uwalniany lokalnie z erytrocytów krwi, proporcjonalnie do spadku wysycenia ich hemoglobiny tlenem (13). W ten sposób, lokalna konsumpcja O2, działająca poprzez lokalne uwalnianie ATP z erytrocytów, staje się regulatorem wysiłkowej hyperemii i tym samym podaży O2 do mięśni. Mechanizm uwalniania ATP ulega upośledzeniu wraz z wiekiem, a także jest mniej aktywny w erytrocytach pochodzących od pacjentów z cukrzycą typu II (9, 14).
4. Wzrost częstości i siły skurczu mięśnia sercowego (poprzez stymulację β-receptorów adrenergicznych), który, wspólnie z mechanizmem Franka-Starlinga, dostosowuje czynność pompy sercowej do zwiększonego powrotu żylnego.
Ostatecznie, podczas wysiłku fizycznego dochodzi do: (a) spadku oporu naczyniowego w pracujących mięśniach i „przekierowania” do nich rzutu minutowego z narządów trzewnych i (b) wzrostu rzutu minutowego serca i VO2, natomiast (c) ciśnienie tętnicze pozostaje na względnie stałym poziomie (tab. 2, ryc. 1). Ta stałość ciśnienia tętniczego (ciśnienia perfuzyjnego), mimo dramatycznego spadku oporu naczyniowego, jest gwarantem prawidłowej perfuzji takich życiowo istotnych narządów jak mózg, serce i mięśnie szkieletowe (tab. 2).
Tabela 2. Rzut minutowy serca, ciśnienie tętnicze, opór naczyniowy i dystrybucja narządowa przepływu krwi w spoczynku i w czasie intensywnego dynamicznego wysiłku fizycznego u wysportowanego młodego mężczyzny.
| Krążenie | Spoczynek | Wysiłek fizyczny |
| ml/min | % | ml/min | % |
Wieńcowe
Mózgowe
Mięśnie szkieletowe
Trzewne
Nerkowe
Skórne
Inne
Pojemność minutowa serca | 250
750
1200
1400
1100
500
600
5800 | 4
13
21
24
19
9
10
100 | 1000
750
22 000
300
900
600
100
25 650 | 4
3
86
1
4
2
0,5
100 |
Średnie ciśnienie tętnicze
Układowy opór naczyniowy | 90 mmHg
15 mmHg min/l | 105 mmHg
4 mmHg min/l |
Zmodyfikowane na podstawie ref. 50.
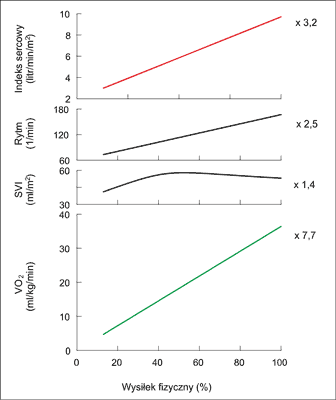
Ryc. 1. Wzrost indeksu sercowego, rytmu serca, rzutu skurczowego serca (SVI) i ustrojowej konsumpcji tlenu (VO2) pod wpływem wzrastającego wysiłku. Dane pochodzą z klasycznego badania Higginbothama i wsp. (49) wykonanego u 24 zdrowych mężczyzn w wieku 20-50 lat ćwiczących na cykloergometrze. Liczby po prawej stronie wykresów określają wielkość rezerwy regulacyjnej danego parametru.
Średnie ciśnienie tętnicze (MAP) jest dane równaniem: MAP = CO x R, gdzie CO oznacza rzut minutowy, a R – całkowity opór naczyniowy. Z pewnym uproszczeniem można przyjąć, że w czasie wysiłku o wielkości ciśnienia tętniczego decydują serce (poprzez rzut minutowy) oraz pracujące mięśnie szkieletowe (poprzez zmianę oporu naczyniowego) (ryc. 2). Aby wysiłek fizyczny nie powodował groźnych spadków ciśnienia tętniczego, rzut minutowy serca musi ściśle nadążać za spadkiem mięśniowego oporu naczyniowego. Szacunki oparte na pomiarach maksymalnych mięśniowych przepływów u wyczynowych sportowców sugerują, że teoretyczny spadek oporu naczyniowego w trakcie pracy mięśniami całego ciała mógłby skutkować przepływem mięśniowym wynoszącym ~100 l/min (9, 11, 12). Oznacza to, że dla utrzymania stałego ciśnienia tętniczego rzut minutowy serca w takich warunkach musiałby mieć podobną wielkość, co prawdopodobnie przekracza możliwości pompy sercowej człowieka. U młodych zdrowych osobników maksymalny rzut minutowy wynosi 20-25 l/min (tab. 2), natomiast największy zanotowany rzut minutowy, zmierzony u wyczynowych sportowców, wynosił nieco ponad 40 l/min (9, 12). Obecnie nie ma pewnej odpowiedzi na pytanie o powód, dla którego rzut minutowy nie wzrasta ponad tę wartość. Wiadomo, że krążenie wieńcowe nie jest czynnikiem limitującym maksymalny rzut minutowy, gdyż maksymalnemu wysiłkowi u zdrowych osób nie towarzyszą objawy niedokrwienia serca (15). Aktualnie dominuje hipoteza, że przyczyna leży po stronie pracujących mięśni limitujących pracę pompy sercowej, a nie po stronie serca. Chodzi o to, że nierównowaga między spadkiem oporu naczyniowego i wzrostem rzutu minutowego musiałaby skutkować spadkiem ciśnienia tętniczego. Prawdopodobnie zapobiega temu odruch z baroreceptorów tętniczych, który poprzez dalszy wzrost aktywności układu współczulnego „dostosowuje” wielkość skurczu naczyń i wielkość oporu naczyniowego w pracujących mięśniach (czynnościowa sympatykoliza ulega osłabieniu) do aktualnej wielkości rzutu minutowego. Umożliwia to utrzymanie stałości ciśnienia tętniczego, ale kosztem ograniczenia perfuzji pracujących mięśni szkieletowych (9-12).
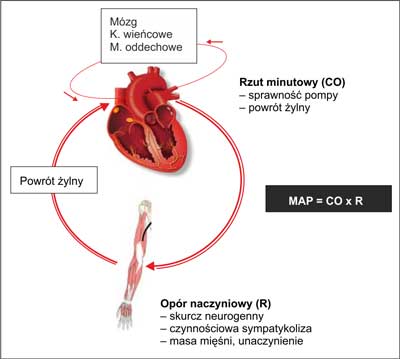
Ryc. 2. Odruchowa regulacja krążenia w czasie wysiłku. Następuje redystrybucja rzutu minutowego serca (CO) z narządów trzewnych do pracujących mięśni. Opór naczyniowy pracujących mięśni determinuje wielkość oporu naczyniowego układu krążenia (R). Układ utrzymuje względnie stałe średnie ciśnienie tętnicze (MAP) (tj. ciśnienie perfuzyjne mózgu, serca i m. oddechowych) poprzez dostosowywanie oporu mięśniowego do rzutu minutowego. Redukcja rzutu minutowego w NS wymusza zwiększony neurogenny skurcz naczyń w pracujących mięśniach.
Przytoczone poniżej przykłady pokazują, że nierównowaga między rzutem minutowym i oporem naczyniowym, skutkująca odruchowym ograniczeniem perfuzji mięśniowej, może być konsekwencją zarówno pracy angażującej zbyt dużą masę mięśni, jak i niesprawności pompy sercowej, jaka towarzyszy NS. Otóż, u wyczynowych sportowców wykazano, że kiedy wykonywali oni maksymalny wysiłek angażujący albo nogi, albo ręce, czynnościowa sympatykoliza i rozkurcz naczyń w aktualnie pracującej kończynie były kompletne. Natomiast jeżeli do maksymalnej pracy nogami dodawali jeszcze intensywną pracę rąk, to przepływ mięśniowy w nogach ulegał redukcji, a przepływ w kończynach górnych ustalał się na poziomie submaksymalnym i miało to związek ze wzrostem uwalniania katecholamin w pracujących kończynach i wzrostem neurogennego przykurczu naczyń (11). W podobnym badaniu u pacjentów z NS (frakcja wyrzucenia 26 ± 9%) wykazano, że praca jedną nogą skutkowała takim samym wzrostem lokalnej mięśniowej perfuzji jak w grupie kontrolnej. Natomiast już dodanie pracy drugą nogą powodowało redukcję o 24% przepływu i o 38% VO2 w pierwszej nodze. Dodatkowo pracy obu kończynami towarzyszył wzrost produkcji katecholamin, jak również niewielki spadek ciśnienia tętniczego (16).
Niemniej jednak, fizjologia wysiłkowej hyperemii w mięśniach szkieletowych jest ciągle słabo poznana. Niekompletna czynnościowa sympatykoliza występuje u wyczynowych sportowców (charakteryzujących się wyjątkowo dużym wysiłkowym przepływem mięśniowym, rzutem minutowym i VO2max) dopiero wtedy, gdy angażują oni mięśnie całego ciała (11, 12). U osób nietrenujących, rezydualny neurogenny skurcz naczyń jest wyraźnie obecny już przy wysiłkach angażujących mniejsze partie mięśni, jest zwiększony u osób nieaktywnych fizycznie, rośnie pod wpływem kilkudniowego unieruchomienia oraz u osób starszych. We wszystkich tych stanach wykazano korelację między osiąganym przepływem mięśniowym oraz maksymalnym rzutem minutowym i VO2max (10, 17, 18). Z drugiej strony, w grupie młodych zdrowych mężczyzn wykazano, że praca kończyną dolną i wazodilatator naczyń mięśniowych (ATP) podany do tętnicy udowej skutkowały podobnym wzrostem rzutu minutowego oraz że izolowane przyspieszenie rytmu serca nie wpływało na wielkość rzutu minutowego ani w spoczynku, ani w czasie pracy czy infuzji ATP (natomiast zmniejszało rzut skurczowy) (19). Badania te pokazują, że: (a) przepływ krwi przez pracujące mięśnie szkieletowe i ich zdolność do pracy są regulowane lokalnie i u zdrowych osobników nie zależą bezpośrednio od serca, (b) ważnym regulatorem rzutu minutowego serca są przepływ mięśniowy/powrót żylny (vide eksperyment z ATP), (c) izolowana modyfikacja czynności pompy sercowej (rytm), bez równoczesnej zmiany powrotu żylnego, pozostaje bez wpływu na rzut minutowy serca.
W skurczowej NS, wraz z postępem choroby od bezobjawowej dysfunkcji lewej komory do NS w klasie NYHA II do IV maleje rezerwa rzutu minutowego, aż do wartości bliskich zeru w krańcowych stadiach NS (ryc. 3). Mimo niewątpliwego upośledzenia czynności pompy sercowej w NS, także i w tym zespole możliwość wykonywania pracy mięśniowej jest bezpośrednio limitowana przez mięśnie, a nie przez serce. Argumenty za tym przemawiające są następujące (5, 6):
1. Nawet u osób z zaawansowaną dysfunkcją serca objawy NS bywają minimalne, a u osób z niewielką dysfunkcją objawy mogą być nasilone.
2. Początkowe obiektywne pogorszenie sprawności hemodynamicznej serca pod wpływem β-blokerów nie skutkuje proporcjonalnym pogorszeniem objawów NS.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. O’Connor CM, Whellan DJ, Lee KL et al.: Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. JAMA 2009; 301: 1439-1450.
2. Piepoli MF, Conraads V, Corra U et al.: Exercise training in heart failure: from theory to practice. A consensus document of the Heart Failure Association and the European Association for Cardiovascular Prevention and Rehabilitation. Eur J Heart Fail 2011; 13: 347-357.
3. Coats AJS: Clinical utility of exercise training in chronic systolic heart failure. Nat Rev Cardiol 2011; 8: 380-392.
4. McMurray JJ, Adamopoulos S, Anker SD et al.: ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2012; 14: 803-869.
5. Clark AL: Origin of symptoms in chronic heart failure. Heart 2006; 92: 12-16.
6. Witte KK, Clark AL: Why does chronic heart failure cause breathlessness and fatigue? Prog Cardiovasc Dis 2007; 49: 366-384.
7. Piepoli M, Crisafulli A: Pathophysiology of human heart failure: importance of skeletal muscle myopathy and reflexes. Exp Physiol 2014; 99(4): 609-615.
8. Boushel R: Muscle metaboreflex control of the circulation during exercise. Acta Physiol (Oxf) 2010; 199: 367-383.
9. Calbet JAL, Lundby C: Skeletal muscle vasodilatation during maximal exercise in health and disease. J Physiol 2012; 590: 6285-6296.
10. Saltin B, Mortensen SP: Inefficient functional sympatholysis is an overlooked cause of malperfusion in contracting skeletal muscle. J Physiol 2012; 590: 6269-6275.
11. Calbet JAL, Jensen-Urstad M, van Hall G et al.: Maximal muscular vascular conductances during whole body upright exercise in humans. J Physiol 2004; 558: 319-331.
12. Saltin B: Exercise hyperaemia: magnitude and aspects on regulation in humans. J Physiol 2007; 583: 819-823.
13. Gonzalez-Alonso J: ATP as a mediator of erythrocyte-dependent regulation of skeletal muscle blood flow and oxygen delivery in humans. J Physiol 2012; 590: 5001-5013.
14. Sprague RS, Bowles EA, Achilleus D, Ellsworth ML: Erythrocytes as controllers of perfusion distribution in the microvasculature of skeletal muscle. Acta Physiol (Oxf) 2011; 202: 285-292.
15. Duncker DJ, Bache RJ: Regulation of coronary blood flow during exercise. Physiol Rev 2008; 88: 1009-1086.
16. Magnusson G, Kaijser L, Sylven C et al.: Peak skeletal muscle perfusion is maintained in patients with chronic heart failure when only a small muscle mass is exercised. Cardiovasc Res 1997; 33: 297-306.
17. Mortensen SP, Nyberg M, Winding K, Saltin B: Lifelong physical activity preserves functional sympatholysis and purinergic signalling in the ageing human leg. J Physiol 2012; 590: 6227-6236.
18. Mortensen SP, Morkeberg J, Thaning P et al.: Two weeks of muscle immobilization impairs functional sympatholysis but increases exercise hyperemia and the vasodilatory responsiveness to infused ATP. Am J Physiol Heart Circ Physiol 2012; 302: H2074-H2082.
19. Bada AA, Svendsen JH, Secher NH et al.: Peripheral vasodilatation determines cardiac output in exercising humans: insight from atrial pacing. J Physiol 2012; 590: 2051-2060.
20. Levine BD: VO2max: what do we know, and what do we still need to know? J Physiol 2008; 586: 25-34.
21. Crisafulli A, Tocco F, Milia R et al.: Progressive improvement in hemodynamic response to muscle metaboreflex in heart transplant recipients. J Appl Physiol (1985) 2013; 114: 421-427.
22. Fulster S, Tacke M, Sandek A et al.: Muscle wasting in patients with chronic heart failure: results from the studies investigating co-morbidities aggravating heart failure (SICA-HF). Eur Heart J 2013; 34: 512-519.
23. Middlekauff HR: Making the case for skeletal myopathy as the major limitation of exercise capacity in heart failure. Circ Heart Fail 2010; 3: 537-546.
24. Rehn TA, Munkvik M, Lunde PK et al.: Intrinsic skeletal muscle alterations in chronic heart failure patients: a disease-specific myopathy or a result of deconditioning? Heart Fail Rev 2012; 17: 421-436.
25. Gielen S, Schuler G, Adams V: Cardiovascular effects of exercise training: molecular mechanisms. Circulation 2010; 122: 1221-1238.
26. Poole DC, Hirai DM, Copp SW, Musch TI: Muscle oxygen transport and utilization in heart failure: implications for exercise (in)tolerance. Am J Physiol Heart Circ Physiol 2012; 302: H1050-H1063.
27. Wang HJ, Li YL, Gao L et al.: Alteration in skeletal muscle afferents in rats with chronic heart failure. J Physiol 2010; 588: 5033-5047.
28. Kato A: Muscle wasting is associated with reduced exercise capacity and advanced disease in patients with chronic heart failure. Future Cardiol 2013; 9: 767-770.
29. Triposkiadis F, Karayannis G, Giamouzis G et al.: The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol 2009; 54: 1747-1762.
30. Floras JS: Sympathetic nervous system activation in human heart failure: clinical implications of an updated model. J Am Coll Cardiol 2009; 54: 375-385.
31. Patel KP, Zheng H: Central neural control of sympathetic nerve activity in heart failure following exercise training. Am J Physiol Heart Circ Physiol 2012; 302: H527-H537.
32. Kishi T: Heart failure as an autonomic nervous system dysfunction. J Cardiol 2012; 59: 117-122.
33. Parati G, Esler M: The human sympathetic nervous system: its relevance in hypertension and heart failure. Eur Heart J 2012; 33: 1058-1066.
34. Sinoway LI, Li J: A perspective on the muscle reflex: implications for congestive heart failure. J Appl Physiol 2005; 99: 5-22.
35. Gao L, Wang W, Liu D, Zucker IH: Exercise training normalizes sympathetic outflow by central antioxidant mechanisms in rabbits with pacing-induced chronic heart failure. Circulation 2007; 115: 3095-3102.
36. Li YL, Gao L, Zucker IH, Schultz HD: NADPH oxidase-derived superoxide anion mediates angiotensin II-enhanced carotid body chemoreceptor sensitivity in heart failure rabbits. Cardiovasc Res 2007; 75: 546-554.
37. Zucker IH: Novel mechanisms of sympathetic regulation in chronic heart failure. Hypertension 2006; 48: 1005-1011.
38. Schultz HD: Nitric oxide regulation of autonomic function in heart failure. Curr Heart Fail Rep 2009; 6: 71-80.
39. Crimi E, Ignarro LJ, Cacciatore F, Napoli C: Mechanisms by which exercise training benefits patients with heart failure. Nat Rev Cardiol 2009; 6: 292-300.
40. Bruno RM, Ghiadoni L, Seravalle G et al.: Sympathetic regulation of vascular function in health and disease. Front Physiol 2012; 3: 284; doi: 10.3389/fphys.2012.00284. eCollection@2012.:284.
41. Davies PF: Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat Clin Pract Cardiovasc Med 2009; 6: 16-26.
42. Sipola P, Heikkinen J, Laaksonen DE, Kettunen R: Influence of 12 weeks of jogging on magnetic resonance-determined left ventricular characteristics in previously sedentary subjects free of cardiovascular disease. Am J Cardiol 2009; 103: 567-571.
43. Taylor RS, Davies EJ, Dalal HM et al.: Effects of exercise training for heart failure with preserved ejection fraction: A systematic review and meta-analysis of comparative studies. Int J Cardiol 2012; 162: 6-13.
44. Esposito F, Reese V, Shabetai R et al.: Isolated quadriceps training increases maximal exercise capacity in chronic heart failure: the role of skeletal muscle convective and diffusive oxygen transport. J Am Coll Cardiol 2011; 58: 1353-1362.
45. Wisloff U, Stoylen A, Loennechen JP et al.: Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation 2007; 115: 3086-3094.
46. Bogdanis GC: Effects of physical activity and inactivity on muscle fatigue. Front Physiol 2012; 3: Article 142.
47. Arena R, Myers J, Forman DE et al.: Should high-intensity-aerobic interval training become the clinical standard in heart failure? Heart Fail Rev 2013; 18: 95-105.
48. Gibala MJ, Little JP, Macdonald MJ, Hawley JA: Physiological adaptations to low-volume, high-intensity interval training in health and disease. J Physiol 2012; 590: 1077-1084.
49. Higginbotham MB, Morris KG, Williams RS et al.: Regulation of stroke volume during submaximal and maximal upright exercise in normal man. Circ Res 1986; 58: 281-291.
50. Harris A, Martin BE: Exercise Physiology. [W:] Rhodes RA, Tanner GA (eds.): Medical Physiology. Lippincott Williams & Wilkins, Philadelphia 2003.
