© Borgis - Postępy Nauk Medycznych 7/2014, s. 487-495
*Urszula Mackiewicz, Joanna Kołodziejczyk, Bohdan Lewartowski
Komórkowe mechanizmy zaburzeń rozkurczu w niewydolności serca
Cellular mechanisms of diastolic dysfunction in the heart failure
Department of Clinical Physiology, Medical Center of Postgraduate Education, Warszawa
Head of Department: prof. Andrzej Beręsewicz, MD, PhD
Streszczenie
Rozkurcz mięśnia sercowego jest złożonym kilkuetapowym procesem. W jego przebiegu wyróżniamy rozkurcz czynny i rozkurcz bierny. Rozkurcz czynny obejmuje usuwanie jonów Ca2+ z cytoplazmy i odłączenie się jonów Ca2+ od aparatu kurczliwego, po którym następuje separacja białek kurczliwych – miozyny i aktyny. Szybkość usuwania jonów Ca2+ z cytoplazmy zależy od aktywności i ekspresji ATP-azy wapniowej siateczki sarkoplazmatycznej oraz zlokalizowanego w błonie komórkowej wymiennika Na+/Ca2+. Z kolei odłączenie Ca2+ od aparatu kurczliwego zależy od wrażliwości troponiny C na jony Ca2+, która jest regulowana przez stopień ufosforylowania troponiny I. Rozkurcz bierny obejmuje przywrócenie długości spoczynkowej kardiomiocytów oraz ich spoczynkowe rozciąganie kardiomiocytów przez napływającą do serca krew. O sprawności tego etapu decydują sztywność białka cytoszkieletu – titiny, zależna od fosforylacji jej sprężystych domen, oraz skład kolagenowy macierzy zewnątrzkomórkowej oplatającej kardiomiocyty. Szczegółowe badania na poziomie komórkowym pokazują, że każdy z wymienionych etapów rozkurczu może być zaburzony w niewydolności serca. Dotyczy to przede wszystkim chorych z rozkurczową postacią niewydolności serca, u których frakcja wyrzucania lewej komory jest zachowana, a objawy są wynikiem zaburzeń rozkurczu. Skuteczna terapia tych pacjentów wymaga znalezienia metod korekty zaburzonych etapów rozkurczu.
Summary
The relaxation of cardiac muscle is the complicated process comprising two stages: active and passive. Active part of relaxation includes ATP-dependent Ca2+ removal from the cytoplasm and dissociation of Ca2+ from contractile apparatus followed by separation of contractile proteins – myosin and actin. Ca2+ removal from the cytoplasm depends on activity and expression of the two main Ca2+ transporters: sarcoplasmic reticulum Ca2+-ATPase and located in extracellular membrane Na+/Ca2+ exchanger. The rate of Ca2+ dissociation from the contractile apparatus is determined by Ca2+ sensitivity of troponin C, which, in turn, depends on the phosphorylation level of troponin I. Passive stage of relaxation process comprises the restoration of cardiomyocyte resting length and subsequent cardiomyocyte stretching through blood flowing into the heart. These processes are determined by the stiffness of titin, the important cytoskeletal protein, as well as by the collagen composition of the extracellular matrix. The titin stiffness is regulated by the phosphorylation level of its elastic domains. Detailed investigations on the cardiomyocyte and the tissue levels reveled disturbances at the each stage of relaxation process in the heart failure patients. It is especially truth for patients with preserved ejection fraction and the diastolic dysfunction as a dominant contributor to symptoms. The effective therapy of these patients requires improvement of diastolic function throughout targeting disturbed stages of the relaxation process.

INTRODUCTION
Heart failure (HF) in the developed countries is diagnosed in 0.2% of population between 35 and 64 years of age. The morbidity increases with age and in the group of people over 80 exceeds 10%. The most frequent cause of HF (about 70% of cases) is ischemic heart disease, including myocardial infarction and hypertension (1-3).
HF and its symptoms, such as dyspnea, exercise intolerance and edema, were for many years connected to impaired systolic function of the left ventricle (LV) and inadequate organ perfusion. Lately, however, it has been highlighted that almost always systolic dysfunction is accompanied by diastolic disturbances. Moreover, in over 50% of patients with symptomatic HF there are only diastolic dysfunction and ejection fraction (EF) is preserved or slightly lowered (EF > 45-50%). The research showed that diastolic dysfunction is more correlated with severity of HF symptoms and patients’ hospitalization than systolic dysfunction. Isolated diastolic dysfunction in HF occur more often in older people, in patients with hypertension, obese patients and patients with type II diabetes (4-6).
Apart from intensive therapy based on multicenter studies long term prognosis in patient with HF is poor (over 50% of patient die in 4 years) (7). Ineffectiveness of treatment concerns, first of all, patients with diastolic dysfunction and preserved EF. In the last few decades survival in this group of patients did not improve (8).
One of the causes of such situation may be the fact that practically in all multicenter trials, testing effectiveness of HF therapy, patients with isolated diastolic dysfunction (FW > 45-50%) were not included in the studies.
Only a few trials were planned for patients with isolated diastolic dysfunction. They revealed that in patients with preserved EF ACE inhibitors, beta-blockers and aldosterone receptor antagonist – spironolacton did not reduce the mortality and HF hospitalization (9-13). On the other hand, statins significantly reduced mortality in patients with HF and preserved EF and did not in patients with lowered EF (14, 15). The only study which showed that the tested intervention was equally effective in patient with preserved or only mildly decreased EF and with significantly lowered EF was the study with third generation beta-blocker – nebivolol (16). The results of the above mentioned studies suggest that treatment bringing benefits to patients with lowered EF is not effective in patient with isolated diastolic dysfunction and that the pathomechanism of these two HF types is different.
The amount of patients with HF will increase due to lengthening of life time and higher survival of patients with myocardial infarction. Due to community ageing and obesity epidemic there will probably be higher percentage of patients with isolated diastolic dysfunction. Finding new effective methods of treatment aimed to improve diastolic function of LV requires deep understanding of the pathomechanism of diastolic disturbances in HF.
During diastole cardiac muscle relax. Cardiac muscle relaxation is a complex multistage process. In its course there is an active and passive component. The active relaxation is an energy consuming process comprising ATP – dependent elimination of Ca2+ from cytoplasm, and Ca2+ disconnecting from the contractile apparatus which results disconnecting between the main contractile proteins – myosin and actin. Passive relaxation involves restitution of resting sarcomere length thanks to elastic recoil generated by cytoskeleton protein titin and resting stretching of cardiomyocytes by blood flowing into the heart. The efficiency of this phase is determined by susceptibility to stretch of the titin and extracellular matrix surrounding cardiomyocytes (fig. 1) (17). Detailed research on the cellular level of heart muscle show that each of the mentioned stages of relaxation process can be disturbed in HF. Modern therapy of patients with HF should be directed to improvement of both relaxation stages.
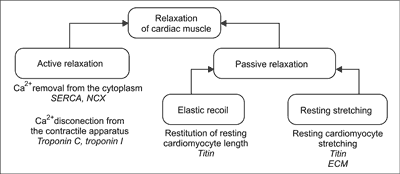
Fig. 1. Stages of cardiac muscle relaxation.
SERCA – sarco-endoplasmic reticulum calcium ATPase; NCX – Na+/Ca2+ exchanger; ECM – extracellular matrix
DISTURBANCES OF ACTIVE RELAXATION PROCESS IN HEART FAILURE
Intracellular Ca2+ handling
The first stage of relaxation process, called active diastole, begins with Ca2+ ions elimination from cytoplasm. Before contraction there is an increase in the concentration of Ca2+ ions in cytoplasm (from about 0.1 μM to 1 μM). This increase is an effect of Ca2+ influx from the extracellular space through voltage activated L type calcium channels. This influx opens calcium channels of the sarcoplasmatic reticulum (SR), also called ryanodine receptors (RyRs). This phenomenon is called calcium induced calcium release (fig. 2). When Ca2+ concentration in cytoplasm increases over 1 μM contractile apparatus protein, troponin C, binds Ca2+ ions. After binding Ca2+ troponin C changes it’s conformation which enables interaction between main contractile proteins: myosin and actin, slipping of actin filaments between myosin ones and as a result cell shortening (contraction) (fig. 2).
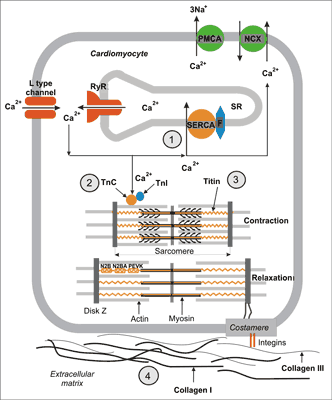
Fig. 2. Cellular mechanisms of cardiac muscle relaxation. After contraction Ca2+ ions are transported to sarcoplasmatic reticulum (SR) by calcium ATPase (SERCA) regulated by fosfolamban (F) and removed to extracellular space by Na+/Ca2+ exchanger (NCX) and membrane ATPase (PMCA) (1). As a result of the decrease of Ca2+ ions concentration in cytoplasm they are released from troponin C (2) which leads to disconnecting of constrictile proteins – actin and myosin. Energy collected in elastic domains (PEVK, N2B, N2BA) of titin molecule generates elastic recoil, which leads to resting sarcomere length restoration (3). Sarcomere stretching over its resting length depends on titin susceptibility to stretch (3) as well as the expression of collagen I and III in the extracellular matrix (4).
Cardiomyocyte relaxation requires Ca2+ removing from cytoplasm. It consecutively enables disconnecting of Ca2+ from troponin C, disconnecting actin and myosin and relaxation. Three ion transporters are responsible for removal of Ca2+ from the cytoplasm after contraction (1) located in SR membranes, sarco-endoplasmatic reticulum calcium ATPase (SERCA), which transports Ca2+ ions to SR, and (2) two proteins located in the cell membrane: Na+/Ca2+ exchanger (NCX) and plasma membrane calcium ATPase (PMCA), which remove Ca2+ ions from the cell (fig. 2) (18).
In human cardiomyocytes SERCA is responsible for removal of about 70-80% of Ca2+ ions from the cytoplasm. That makes it a protein which determines the rate of active stage of relaxation process at the highest degree. Transport of Ca2+ ions to SR is energy consuming. SERCA uses about 15% of cardiomyocyte ATP. SERCA is inhibited by the protein localized in SR membranes – phospholamban. Phospholamban phosphorylation decrease SERCA inhibition. Phospholamban is phosphorylated by two kinases: protein kinase A (PKA) activated by stimulation of β-1 adrenergic receptors and by Ca2+ and calmodulin dependent kinase (CAMKII), activated by increase in the heart rate (which means also under catecholamine stimulation). In turn phosphatases PP1, PP2A and PP2B (calcineurin) are responsible for phospholamban dephosphorylation. Stimulation of Gq protein coupled receptors (such as angiotensin II or endothelin-1 receptors) or increase in wall stress in the heart muscle activates these phosphatases (19). The final level of phospholamban phosphorylation (and SERCA activity) depends on the resultant of kinases and phosphatases activity.
NCX localized in the cell membrane is the main route of Ca2+ eflux. It removes about 20% of Ca2+ ions activating contraction (PMCA is responsible for removal of about 1-4% of Ca2+ ions). NCX is not able to utilize ATP. It takes the energy required to Ca2+ transport from sodium gradient (during one Ca2+ ion removal from the cell three Na+ ions are transported into the cell) (18). All factors leading to lowering transmembrane sodium gradient (such as inhibition of Na+/K+ pump by digitalis glycosides or ion imbalances like hypokalaemia or hypomagnesaemia inhibit NCX activity and lead to intracellular Ca2+ accumulation).
SERCA activity and at a lower degree NCX determine the rate of Ca2+ ions removal from cytoplasm and thus the rate of their disconnecting from troponin C. This last process is additionally regulated by troponin C sensitivity to Ca2+ ions, which depends on the level of troponin I phosphorylation. This subunit of troponin is phosphorylated by three kinases: by PKA (similarly as phospholamban), by activated by nitric oxide (NO) and natriuretic peptydes protein kinase G (PKG) and by protein kinase C (PKC) activated under activation of Gq protein coupled receptors troponin I phosphorylation by PKA and PKG lowers troponin C sensitivity to Ca2+ ions which makes relaxation easier. On the other hand, phosphorylation by PKC has a reverse effect (20).
Summing up, the rate of the active part of relaxation depends on the rate of Ca2+ ions removal from cytoplasm and on the rate of Ca2+ ions disconnecting from troponin C. The first process mostly depends on SERCA activity regulated by the level of phospholamban phosphorylation. The second one depends on the troponin C Ca2+ sensitivity, which is regulated by the level of troponin I phosphorylation.
Neurohumoral regulation of intracellular Ca2+ handling
Regulation of intracellular Ca2+ handling and thus cardiomyocytes contraction-relaxation cycle by the sympathetic nervous system is, beside Frank-Starling mechanism, a main mechanism adjusting heart muscle to increased work in conditions of physical and psychical stress (fight or flight response). Activation of sympathetic system causes noradrenalin release from the sympathetic nerve terminals and adrenalin from the medulla of adrenal glands. Catecholamines, by activating beta-adrenergic receptors (mainly beta-1), lead to, in turn, activation of Gs protein, adenylate cyclase synthetizing cAMP and to activation of cAMP-dependent PKA. In addition, catecholamines cause increase in the heart rate which leads to CaMKII activation (21).
Activation of PKA and CaMKII by phosphorylation of a few Ca2+ handling proteins leads to increase in contraction strength as well as the rate of cardiomyocytes relaxation. The objects of phosphorylation are: L type Ca2+ channels, RyRs, phospholamban and troponin I.
Phosphorylation of L type Ca2+ channels increases Ca2+ influx and the amount of Ca2+ ions released from SR. RyRs phosphorylation increases their sensitivity to Ca2+ and makes their opening easier. Phospholamban phosphorylation increases SERCA activity, Ca2+ ions transport to SR and the amount of Ca2+ collected in the SR. Troponin I phosphorylation decreases troponin C sensitivity to Ca2+ ions and makes their disconnecting from protein easier, which promote relaxation.
Higher Ca2+ influx, higher Ca2+ sensitivity of RyRs and higher activity of SERCA are the main elements of positive inotropic catecholamine effect. Increased SERCA activity and decrease in troponin C Ca2+ sensitivity make relaxation easier and are the main elements of positive lusitropic catecholamine effect (22).
Ca2+ handling disturbances in the heart failure
In HF, in animal models as well as in human explanted hearts, there are numerous disturbances of expression and function of proteins engaged in intracellular Ca2+ handling. The most frequent observation is the decrease in expression and activation of SERCA, excessive increase in RyRs Ca2+ sensitivity to and increase in NCX expression.
Decrease in SERCA expression in explanted hearts may even reach 50%. Moreover, there is also a decrease level of phospholamban phosphorylation and thus higher inhibition of SERCA (23, 24). Lowered level of phospholamban phosphorylation in HF is not fully understood. On the one hand, due to intensive catecholamine stimulation activation of PKA and CaMKII kinases increases, which should lead to increase in phospholamban phosphorylation. On the other hand, however, the increase in the level of angiotensin II results in AT1 receptors activation and subsequent PKC activation which is the main activator of PP1 phosphatase. Additionally, in HF (particularly in its diastolic form) activity of PP2B phosphatase (calcineurin) increases (25, 26). It seems that the resultant effect is the predominance of phosphatases effect over kinase, which leads to decrease in phospholamban phosphorylation, decrease in SERCA activity and finally lowering the rate of relaxation. In addition, the decrease in SE CA expression and activity leads to lowering of the SERCA2+ content and to the decrease in contraction amplitude.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Cowie MR, Mostead A, Wood DA et al.: The epidemiology of heart failure. Eur Heart J 1997; 18: 208-225.
2. Dickstein K, Cohen-Solal A, Filippatos G et al.: ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J 2008; 29: 2388-2442.
3. Hole T, Hall C, Skjaerpe T: N-terminal proatrial natriuretic peptide predicts two-year remodelling in patients with acute transmural myocardial infarction. Eur Heart J 2004; 25: 416-423.
4. Paulus WJ, Tschope C, Sanderson JE et al.: How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J 2007; 28: 2539-2550.
5. Gulec S, Ertas F, Tutar E et al.: Exercise performance in patients with dilated cardiomyopathy: relationship to resting left ventricular function. Int J Cardiol 1998; 65: 247-253.
6. Skaluba SJ, Litwin SE: Mechanisms of exercise intolerance: insights from tissue Doppler imaging. Circulation 2004; 109: 972-977.
7. Mosterd A, Hoes AW: Clinical epidemiology of heart failure. Heart 2007; 93: 1137-1146.
8. Owan TE, Hodge DO, Herges RM et al.: Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 2006; 355: 251-259.
9. Yusuf S, Pfeffer MA, Swedberg K et al.: Effects of candesartan in patients with chronic hear failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362: 777-781.
10. Cleland JG, Tendera M, Adamus J et al.: Perindopril for elderly people with chronic hearnfailure: the PEP-CHF study. The PEP investigators. Eur J Heart Fail 1999; 1: 211-217.
11. Lam CS, Carson PE, Anand IS et al.: Sex differences in clinical characteristics and outcomes in elderly patients with heart failure and preserved ejection fraction: the Irbesartan in Heart Failure with Preserved Ejection Fraction (I-PRESERVE) trial. Circ Heart Fail 2012; 5: 571-578.
12. Hernandez AF, Hammill BG, O’Connor CM et al.: Clinical effectiveness of beta-blockers in heart failure: findings from the OPTIMIZE-HF (Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure) Registry. J Am Coll Cardiol 2009 Jan 13; 53(2): 184-192.
13. Fukuta H, Sane DC, Brucks S, Littre WC: Statin Therapy May Be Associated With Lower Mortality in Patients With Diastolic Heart Failure: a preliminary raport. Circulation 2005; 112: 357-363.
14. Kjekshus J, Apetrei E, Barrios V et al.: Rosuvastatin in older patients with systolic heart failure. N Engl J Med 2007; 357: 2248-2261.
15. Edelmann F, Gelbrich G, Duvinage A et al.: Differential interaction of clinical characteristics with key functional parameters in heart failure with preserved ejection fraction – Results of the Aldo-DHF trial. Int J Cardiol 2013 Nov 30; 169(6): 408-417.
16. Ghio S, Magrini G, Serio A et al.: Effects of nebivolol in elderly heart failure patients with or without systolic left ventricular dysfunction: results of the SENIORS echocardiographic sub-placebo-controlled crossover study. Eur Heart J 2006; 27(5): 562-568.
17. Opie LH: Mechanisms of Cardiac Contraction and Relaxation. Braunwald’s Heart Disease: A Textbook of Cardiovascular Medicine. 8th ed., Saunders Elsevier 2007.
18. Bers DM, Gua T: Calcium signaling in cardiac ventricular myocytes. Ann NY Acad Sci 2005: 1047: 86-98.
19. Neumann J, Eschenhagen T, Jones LR et al.: Increased expression of cardiac phosphatases in patients with end-stage heart failure. J Mol Cell Cardiol 1997; 29: 265-272.
20. Marston SB, de Tombe PP: Troponin phosphorylation and myofilament Ca2+-sensitivity in heart failure: increased or decreased? J Mol Cell Cardiol 2008; 45: 603-607.
21. Mackiewicz U, Klemenska E, Beręsewicz A: Receptory beta-adrenergiczne w zdrowym i niewydolnym sercu. Kardiologia Polska 2007; 65: 294-302.
22. Lohse MJ, Engelhardt S, Eschenhagen T: What is the role of beta-adrenergic signaling in heart failure? Circ Res 2003; 93: 896-906.
23. Hasenfuss G, Pieske B: Calcium cycling in congestive heart failure. J Mol Cell Cardiol 2002; 34: 951-969.
24. Yano M, Ikeda Y, Matsuzaki M: Altered intracellular Ca2+ handling in hart failure. J Clin Invest 2005; 115: 556-564.
25. Huang B, Wang S, Qin D et al.: Diminished basal phosphorylation level of phospholamban in the postinfarction remodeled rat ventricle: role of beta-adrenergic pathway, G(i) protein, phosphodiesterase, phosphatases. Circ Res 1999; 85: 848-855.
26. Gupta RC, Mishra S, Rastogi S et al.: Cardiac SR-coupled PP1 activity and expression are increased and inhibitor 1 protein expression is decreased in failing hearts. Am J Physiol Heart Circ Physiol 2003; 285: H2373-H2381.
27. Marx SO, Marks AR: Dysfunctional ryanodine receptors in the heart: new insights into complex cardiovascular diseases. Mol Cell Cardiol 2013; 58: 225-231.
28. Nattel S, Maguy A, LeBouter S, Yeh Y-H: Arrhythmogenic ion-channel remodeling in the heart: heart failure, myocardial infarction, and atrial fibrillation. Physiol Rev 2007; 87: 425-456.
29. Marx SO, Reiken S, Hisamatsu Y et al.: PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell 2000; 101: 365-376.
30. Mackiewicz U, Lewartowski B: Temperature dependent contribution of Ca2+ transporters to relaxation in cardiac myocytes: important role of sarcolemmal Ca2+-ATPase. J Physiol Pharmacol 2006; 57: 3-15.
31. Rundqvist B, Elam M, Bergmann-Sverrisdottir Y et al.: Increased cardiac adrenergic drive precedes generalized sympathetic activation in human heart failure. Circulation 1997 Jan 7; 95(1): 169-175.
32. Marks AR, Reiken S, Marx SO: Progression of heart failure: is protein kinase a hyperphosphorylation of the ryanodine receptor a contributing factor? Circulation 2002; 105(3): 272-275.
33. Ai X, Curran JW, Shannon TR et al.: Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res 2005; 97: 1314-1322.
34. Sun YL, Hu SJ, Wang LH et al.: Comparison of low and high doses of carvedilol on restoration of cardiac function and calcium-handling proteins in rat failing heart. Clin Exp Pharmacol Physiol 2005 Jul; 32(7): 553-560.
35. Schillinger W, Schneider H, Minami K et al.: Importance of sympathetic activation for the expression of Na+-Ca2+ exchanger in end-stage failing human myocardium. Eur Heart J 2002 Jul; 23(14): 1118-1124.
36. van Heerebeek L, Hamdani N, Falcão-Pires I et al.: Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 2012; 126(7): 830-839.
37. Hamdani N, Bishu KG, von Frieling-Salewsky M et al.: Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res 2013; 97(3): 464-471.
38. Nawarskas JJ, Anderson JR: Levosimendan: a unique approach to the treatment of heart failure. Heart Dis 2002; 4(4): 265-271.
39. LeWinter MM, Granzier H: Cardiac Titin – A Multifunctional Giant. Circulation 2010 May 18; 121(19): 2137-2145.
40. Martin M, LeWinter MD, Granzier HL: Cardiac Titin and Heart Disease. Journal of Cardiovascular Pharmacology 2014 March; 63(3): 207-212.
41. Granzier HL, Irving TC: Passive tension in cardiac muscle: contribution of collagen, titin, microtubules and intermediate filaments. Biophys J 1996; 68: 1027-1044.
42. Hidalgo CG, Chung CS, Saripalli C et al.: The multifunctional Ca2+/calmodulin-dependent proteinkinase II delta (CaMKII phosphorylates cardiac titin’s spring elemets. J Mol Cell Cardiol 2013; 54: 90-97.
43. Neagoe C, Kulke M, del Monte F et al.: Titin isoform switch in ischemic human heart disease. Circulation 2002; 106: 1333-1341.
44. Nagueh SF, Shah G, Wu Y et al.: Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004; 110: 155-162.
45. van Heerebeck L, Borbely A, Nissen HW et al.: Myocardial structure and function differ in systolic and diastolic heart failure. Circulation 2006; 113: 1966-1973.
46. Borbely A, van der Velden J, Papp Z et al.: Cardiomyocyte stiffness in diastolic heart failure. Circulation 2005; 111: 774-781.
47. Hamdani N, Krysiak J, Kreussner MM et al.: Crucial role for Ca2+/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res 2013 Jan, ahead of print.
48. van Heerebeek L, Franssen CPM, Hamdani N et al.: Molecular and cellular basis for diastolic dysfunction. Curr Heart Fail Rep 2012; 9: 293-302.
49. Takimoto E: Cyclic GMP-dependent signaling in cardiac myocytes. Circulation J 2012; 76: 1819-1825.
50. Guazzi M, Vincenzi M, Arena R, Guazzi MD: Pulmonary hypertension in heart failure with preserved ejection raction: a target of phosphodisterase-5 inhibition in a 1-year study. Circulation 2011a; 124: 164-174.
51. Redfield MM, Chen HH, Borlaug BA et al.: Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction. A randomized clinical trial. JAMA 2013; 309: 1268-1277.
52. Clark KA, McElhinny AS, Beckerle MC, Gregorio CC: Striated muscle cytoarchitecture: an intricate web of form and function. Annu Rev Cell Dev Biol 2002; 18: 637-706.
53. LeGrice I, Pope A, Smaill B: The architecture of the heart: myocyte organization and the cardiac extracellular matrix. [In:] Villarreal FJ (ed.): Interstitial fibrosis in heart failure. NY: Springer, New York 2005: 5-21.
54. Spinale FG, Janicki JS, Zile MR: Membrane-Associated Matrix Proteolysis and Heart Failure. Circ Res 2013; 112: 195-208.
55. Marchesia C, Dentalia F, Nicolinia E et al.: Plasma levels of matrix metalloproteinases and their inhibitors in hypertension: a systematic review and meta-analysis. J Hypertension 2012; 30: 3-16.
56. Polyakova V, Hein S, Kostin S et al.: Matrix metalloproteinases and their tissue inhibitors in pressure-overloaded human myocardium during heart failure progression. J Am Coll Cardiol 2004; 44(8): 1609-1618.
57. Fielitz J, Leuschner M, Zurbrügg HR et al.: Regulation of matrix metalloproteinases and their inhibitors in the left ventricular myocardium of patients with aortic stenosis. J Mol Med 2004; 82(12): 809-820.
58. López B, González A, Querejeta R et al.: Alterations in the pattern of collagen deposition may contribute to the deterioration of systolic function in hypertensive patients with heart failure. J Am Coll Cardiol 2006 Jul 4; 48(1): 89-96. Epub 2006 Jun 12.
59. Mann DL, Spinale FG: Activation of matrix metalloproteinases in the failing human heart: breaking the tie that binds. Circulation 1998; 98(17): 1699-1702.
60. Reiken S, Wehrens XH, Vest JA et al.: Beta-blockers restore calcium release channel function and improve cardiac muscle performance in human heart failure. Circulation 2003; 107(19): 2459-2466.
61. Doi M, Yano M, Kobayashi S et al.: Propranolol prevents the development of heart failure by restoring FKBP12.6-mediated stabilization of ryanodine receptor. Circulation 2002; 105(11): 1374-1379.
62. Satoh S, Ueda Y, Suematsu N et al.: Beneficial effects of angiotensin-converting enzyme inhibition on sarcoplasmic reticulum function in the failing heart of the Dahl rat. Circ J 2003; 67(8): 705-711.
63. Lijnen PJ, Petrov VV: Role of intracardiac renin-angiotensin-aldosterone system in extracellular matrix remodeling. Methods Find Exp Clin Pharmacol 2003; 25(7): 541-564.
64. Shah SJ, Blair JE, Filippatos GS et al.: Effects of istaroxime on diastolic stiffness in acute heart failure syndromes: results from the Hemodynamic, Echocardiographic, and Neurohormonal Effects of Istaroxime, a Novel Intravenous Inotropic and Lusitropic Agent: a Randomized Controlled Trial in Patients Hospitalized with Heart Failure (HORIZON-HF) trial. Am Heart J 2009; 157: 1035-1041.
65. Jessup M, Greenberg B, Mancini D et al.: Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID) Investigators.Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation 2011 Jul 19; 124(3): 304-313.
66. Pleger ST, Shan C, Ksienzyk J et al.: Cardiac AAV9-S100A1 gene therapy rescues post-ischemic heart failure in a preclinical large animal model. Sci Transl Med 2011; 3(92): 92-104.
67. Tilemann L, Lee A, Ishikawa K et al.: SUMO-1 Gene Transfer Improves Cardiac Function in a Large-Animal Model of Heart Failure. Sci Transl Med 2013, in press.
