© Borgis - Anestezjologia Intensywna Terapia 2/2003, s. 126-131
Jerzy Windyga
Rozsiane krzepnięcie śródnaczyniowe w przebiegu posocznicy: patofizjologia, rozpoznawanie i leczenie
Sepsis-induced disseminated intravascular coagulation: pathophysiology, diagnosis and management. A review
Samodzielna Pracownia Krzepnięcia Krwi i Hemostazy Instytutu Hematologii i Transfuzjologii w Warszawie;
Kierownik: prof. dr hab. med. S. Łopaciuk

Wstęp
Rozsiane krzepnięcie śródnaczyniowe ( disseminated intravascular coagulation, DIC) nie jest odrębną jednostką chorobową, lecz zespołem wtórnym do wielu różnych chorób i stanów klinicznych. Jego istotą jest aktywacja procesu krzepnięcia krwi połączona z uczynnieniem lub zahamowaniem fibrynolizy. W następstwie aktywacji krzepnięcia w mikrokrążeniu i – rzadziej – w obrębie wielkich naczyń powstają mnogie zakrzepy, które są przyczyną niedokrwiennego uszkodzenia wielu narządów. Krwinki czerwone uwięzione w sieci włóknika i przepływające przez zmieniony obszar mikrokrążenia ulegają uszkodzeniu i hemolizie. Jednocześnie dochodzi do zużycia płytkowych krwinek, fibrynogenu i innych czynników krzepnięcia. Niedobór tych składników w krążącej krwi objawia się skazą krwotoczną.
Jedną z najczęstszych przyczyn DIC jest sepsa. Jest ona definiowana jako współistnienie zespołu systemowej odpowiedzi zapalnej ( systemic inflammatory response syndrome, SIRS) i zakażenia (tab. I). W osoczu wszystkich pacjentów z ciężką sepsą wykrywa się w osoczu markery aktywacji krzepnięcia krwi, a w około 30-50% przypadków rozpoznaje ostry zespół DIC (1, 2, 3). W ostatnich latach udało się w znacznym stopniu wyjaśnić, w jaki sposób w przebiegu ciężkich zakażeń dochodzi do uogólnionej aktywacji krzepnięcia krwi. Jednocześnie zaproponowano nowe sposoby leczenia pacjentów z ciężką sepsą.
Tabela I. Kryteria rozpoznania zespołu systemowej odpowiedzi zapalnej, posocznicy i zespołu niewydolności wielonarządowej (wg Bone´a i wsp. [26])
| Stan kliniczny | Kryteria rozpoznania |
SIRS
Posocznica
Ciężka posocznica
MODS | Wystąpienie ł 2 spośród następujących objawów:
mTemp. > 38°C lub < 36°C
mTachykardia > 90 /min
mTachypnoe > 20 /min lub PaCO2 < 4,3 kPa
mWBC > 12 000 /mm3 lub < 4000 /mm3 lub obecność
ł 10% granulocytów pałeczkowatych we wzorze odsetkowym leukocytów krwi obwodowej
SIRS + udokumentowane zakażenie
SIRS + udokumentowane zakażenie + zaburzenia hemodynamiczne
Niewydolność wielu narządów skutkująca zaburzeniem homeostazy |
Aktywacja krzepnięcia krwi i zahamowanie fibrynolizy
W badaniach na zdrowych ochotnikach wykazano, że dożylne wstrzyknięcie małej dawki endotoksyny (2-4 ng kg-1), będącej składnikiem otoczki bakterii Gram-ujemnych, powoduje aktywację krzepnięcia krwi (4). Wyrazem tej aktywacji jest zwiększenie zawartości fibrynopeptydu A (FPA), fragmentu 1+2 protrombiny (F 1+2) i kompleksów trombina-antytrombina (TAT) w osoczu. Pojawienie się markerów aktywacji krzepnięcia występuje już w 2 h po podaniu endotoksyny i utrzymuje przez 6-12 h. Pobudzeniu krzepnięcia towarzyszy krótkotrwałe i przelotne uczynnienie fibrynolizy przez uwalniany ze śródbłonka naczyniowego tkankowy aktywator plazminogenu (t-PA). W osoczu stwierdza się wówczas obecność kompleksów plazmina-alfa2-antyplazmina (PAP) oraz produktów degradacji fibrynogenu i fibryny (FDP) i D-dimerów. Później następuje uwalnianie do osocza inhibitorów aktywatorów plazminogenu (PAI), oraz nasila się aktywacja inhibitora fibrynolizy aktywowanego przez trombinę ( thrombin activatable fibrinolysis inhibitor, TAFI), co prowadzi do zahamowania układu fibrynolitycznego na około 6-12 h. Podobną sekwencję wydarzeń opisano u pawianów, którym wstrzyknięto letalną dawkę żywych kolonii Escherichia coli. Opisane zachwianie równowagi pomiędzy układami krzepnięcia i fibrynolizy prowadzi do odkładania i przetrwania złogów fibryny w mikrokrążeniu.
Obecnie ugruntowany jest pogląd, że aktywację krzepnięcia krwi w przebiegu posocznicy inicjuje kompleks czynnika tkankowego ( tissue factor, TF) z aktywnym czynnikiem VII (VIIa) (5). Czynnik tkankowy jest glikoproteiną (47 kDa), stanowiącą integralny składnik błon wielu komórek. TF jest stale syntetyzowany i eksponowany na powierzchni komórek podśródbłonkowej tkanki łącznej, błony środkowej i przydanki naczyń, odsłanianych w uszkodzonej ścianie naczyniowej. W warunkach fizjologicznych czynnik tkankowy nie jest eksponowany na powierzchni komórek śródbłonka. Jednakże pod wpływem pewnych bodźców, np. endotoksyny bakterii Gram-ujemnych, komórki śródbłonka i monocyty są pobudzane do syntezy i ekspresji powierzchniowej TF. Przyłączenie do TF czynnika VIIa i powstanie kompleksu TF-VIIa uruchamia kaskadę krzepnięcia w szlaku zewnątrzpochodnym. O tym, że powstanie kompleksu TF-VIIa odgrywa istotną rolę w rozwoju DIC na tle posocznicy świadczą wyniki badań przeprowadzonych na zwierzętach. Zastosowanie przeciwciał przeciwko czynnikowi VIIa lub przeciwko TF hamowało bowiem aktywację krzepnięcia krwi u szympansów, którym wstrzyknięto endotoksynę (4).
W posocznicy obserwowano aktywację czynników kontaktu, uważaną za mechanizm zapłonowy krzepnięcia w szlaku wewnątrzpochodnym. O aktywacji tej świadczy obniżenie stężenia czynnika XII i prekalikreiny w osoczu. Zastosowanie przeciwciał przeciwko aktywnemu czynnikowi XII nie wpłynęło jednak na zahamowanie generacji trombiny w doświadczalnym DIC. Dlatego uważa się, że aktywacja czynników kontaktu przez bodźce prowokujące DIC nie ma znaczenia dla aktywacji krzepnięcia (5).
Rola cytokin
Cytokiny są białkami o masie cząsteczkowej 8-30 kD, syntetyzowanymi i uwalnianymi przez różne komórki (leukocyty, fibroblasty, komórki śródbłonka i inne) pod wpływem wielu bodźców. Uczestniczą one w wielu ważnych dla ustroju procesach biologicznych, m.in. inicjują i regulują zjawiska immunozapalne w przebiegu zakażeń (6). Pod wpływem endotoksyny bakterii Gram-ujemnych lub egzotoksyny bakterii Gram-dodatnich, makrofagi tkankowe syntetyzują i uwalniają różne cytokiny, w tym czynnik martwicy nowotworów ( tumor necrosis factor, TNF) oraz interleukiny 1, 6 i 8 (IL-1, IL-6, IL-8) (7). Cytokiny te odgrywają bardzo ważną rolę w miejscowych reakcjach obrony ustroju przed zakażeniami, gdyż przyciągają granulocyty do ogniska infekcji. Jednakże uwolnienie dużych ilości tych cytokin do krwiobiegu – zjawisko obserwowane w przebiegu ciężkich posocznic – doprowadza do uszkodzenia układu sercowo-naczyniowego i narządów wewnętrznych (8). Przyczyn tego uszkodzenia upatruje się zarówno w efektach prozapalnych cytokin, jak i w ich wpływie na układy krzepnięcia krwi i fibrynolizy.
Cytokiny uczestniczące w reakcjach zapalnych można podzielić na prozapalne – wykazujące właściwości prozakrzepowe, i przeciwzapalne o właściwościach przeciwstawnych (6, 9). Do cytokin prozapalnych zalicza się TNF oraz IL-1, IL-6, IL-8, zaś do przeciwzapalnych – interleukiny 4, 10 i 13 (IL-4, IL-10, IL-13). TNF i IL-1 wywołują wzrost ciepłoty ciała, spadek ciśnienia tętniczego krwi, bóle mięśniowe i senność. Cytokiny te indukują syntezę i ekspresję białek adhezyjnych na powierzchni komórek krwi i śródbłonka, co sprzyja powstawaniu agregatów komórkowych zamykających światło małych naczyń. Istotnym efektem obu cytokin jest inicjowanie kolagenolizy uszkadzającej ścianę naczyniową. IL-6 stymuluje syntezę IgG i białek ostrej fazy, a także pobudza proliferację megakariocytów i produkcję płytek krwi. IL-8 wywiera działanie angiogenne oraz jest chemoatraktanem dla leukocytów. Cytokiny przeciwzapalne hamują cytotoksyczne działanie monocytów i makrofagów oraz syntezę cytokin prozapalnych i białek ostrej fazy.
Wyniki badań ostatnich lat w dużym stopniu wyjaśniły udział cytokin w zaburzeniach hemostazy prowadzących do DIC (ryc. 1). Hemostatyczne efekty cytokin badano in vivo i in vitro. W badaniach in vivo śledzono skutki dożylnego wstrzyknięcia rekombinowanego TNF, rekombinowanej IL-6 oraz IL-1 (10, 11). Cytokiny te podawano zdrowym ochotnikom, chorym z zaawansowanymi nowotworami złośliwymi oraz szympansom. Wykazano, że użyte w badaniu cytokiny aktywują krzepnięcie krwi oraz – po krótkotrwałym uczynnieniu – hamują układ fibrynolityczny. Najsilniejszym mediatorem aktywacji krzepnięcia krwi jest IL-6, natomiast TNF przede wszystkim tłumi aktywność układu fibrynolizy (10). Bardzo interesujące wyniki przyniosły badania in vitro z zastosowaniem hodowli komórkowych oraz izolowanych naczyń. Wykazano w nich, że prozapalne cytokiny stymulują syntezę i ekspresję powierzchniową TF w monocytach i makrofagach oraz śródbłonku naczyń krwionośnych (5). Jak już wcześniej wspomniano, jest to najważniejszy mechanizm indukcji DIC w przebiegu posocznicy.
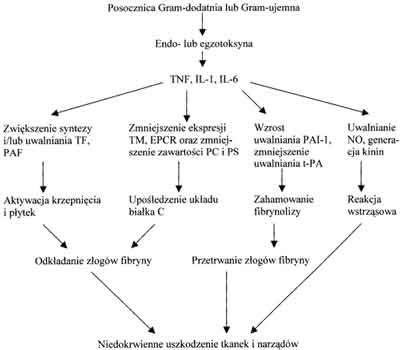
Ryc. 1. Rola prozapalnych cytokin w patogenezie rozsianego krzepnięcia śródnaczyniowego; TNF – czynnik martwicy nowotworów, IL-1 – interleukina 1, IL-6 – interleukina 6, TF – czynnik tkankowy, PAF – czynnik aktywujący płytki, TM – trombomodulina, EPCR – śródbłonkowy receptor dla białka C, PC – białko C, PS – białko S, PAI-1 – inhibitor aktywatora plazminogenu typu 1, t-PA – tkankowy aktywator plazminogenu, NO – tlenek azotu
Wpływ cytokin na hemostazę nie ogranicza się do oddziaływań z układem krzepnięcia krwi i fibrynolizy (5, 9). Prozapalne cytokiny aktywują krwinki płytkowe i wywołują ich agregację. Przyczyniają się także do aktywacji granulocytów. Aktywowane granulocyty zawierają na swej powierzchni białka adhezyjne oraz wytwarzają i uwalniają do krwiobiegu różne substancje, w tym pochodne kwasu arachidonowego, wolne rodniki, czynnik aktywujący płytki, cytokiny oraz enzymy – elastazę i katepsynę G. Ten ostatni enzym aktywuje i agreguje płytki tak samo skutecznie jak trombina. TNF i IL-1 indukują uwalnianie ze śródbłonka naczyniowego tlenku azotu (NO). Szybkie uwolnienie dużych ilości NO jest odpowiedzialne za gwałtowny spadek ciśnienia tętniczego krwi (reakcja wstrząsowa).
Cytokiny wywierają wpływ na reakcje hemostatyczne, natomiast poszczególne elementy hemostazy, a więc składowe układów krzepnięcia krwi i fibrynolizy, płytki krwi oraz ściana naczyń krwionośnych, wpływają na aktywność cytokin. Na przykład trombina stymuluje ekspresję IL-6 w monocytach i śródbłonku naczyniowym, a fibryna zwiększa ekspresję IL-1 w leukocytach. Powstaje błędne koło, w którym prozapalne cytokiny aktywują krzepnięcie, które z kolei indukuje generację cytokin (5).
Endogenne mechanizmy ograniczające krzepnięcie krwi
W przebiegu ciężkiej posocznicy dochodzi zazwyczaj do niedoboru endogennych inhibitorów krzepnięcia, wśród których najważniejszą rolę przypisuje się antytrombinie (dawna nazwa antytrombina III), układowi inhibitorowemu białka C i inhibitorowi zewnątrzpochodnego szlaku krzepnięcia ( tissue factor pathway inhibitor, TFPI) (12, 13).
Antytrombina jest glikoproteiną inaktywującą proteazy serynowe, w tym trombinę. W stanach przebiegających ze wzmożoną generacją trombiny, np. w DIC na tle posocznicy, zawartość antytrombiny w osoczu zmniejsza się, gdyż jest ona zużywana do unieczynnienia trombiny. Antytrombina wykazuje też właściwości przeciwzapalne. Wiążąc się z glikozaminoglikanami (GAG) zlokalizowanymi na powierzchni śródbłonka naczyniowego, antytrombina stymuluje komórki śródbłonka do uwalniania prostacykliny (PGI2), która upośledza agregację płytek krwi oraz hamuje uwalnianie TNF z zaktywowanych monocytów i rodników tlenowych z pobudzonych granulocytów. Warto podkreślić, że region w cząsteczce antytrombiny odpowiedzialny za wiązanie z GAG jest tym samym regionem, który wiąże się z heparyną. Oznacza to, że antytrombina związana z heparyną nie wykazuje właściwości przeciwzapalnych (14).
W skład antykoagulacyjnego układu białka C wchodzi białko C (PC), śródbłonkowy receptor dla białka C (EPCR), białko S (PS) i trombomodulina (TM). W wyniku oddziaływań PC z kompleksem utworzonym przez trombinę i TM, powstaje aktywowane białko C (APC). Zadaniem APC jest proteolityczna degradacja czynników krzepnięcia Va i VIIIa. Degradacja ta zachodzi w obecności wolnego białka S jako kofaktora, fosfolipidów i jonów wapnia. W przebiegu posocznicy dochodzi do upośledzenia układu białka C z powodu hamującego wpływu prozapalnych cytokin na syntezę i ekspresję trombomoduliny, EPCR oraz białek C i S (12, 13).
Inhibitor zewnątrzpochodnego szlaku krzepnięcia jest białkiem syntetyzowanym w wątrobie, śródbłonku naczyniowym i megakariocytach. TFPI inaktywuje kompleks TF-VIIa w obecności aktywnego czynnika X (Xa). Dożylne wstrzyknięcia rekombinowanego TFPI skutecznie hamowały generację trombiny w krwiobiegu zdrowych ochotników, którym uprzednio podano niewielkie dawki endotoksyny (12). Obserwacja ta budzi nadzieje na wykorzystanie TFPI do leczenia chorych z posocznicą, choć należy dodać, że stężenie tego inhibitora w osoczu pozostaje w zakresie normy nawet w DIC o ostrym przebiegu (15).
Rozpoznawanie DIC
Rozsiane krzepnięcie śródnaczyniowe w przebiegu posocznicy ma przebieg ostry. Dominującym objawem klinicznym jest skaza krwotoczna. Jej przyczyn upatruje się w zużyciu płytek krwi i czynników krzepnięcia, a także w zaburzeniach agregacji płytek krwi oraz upośledzeniu polimeryzacji monomerów fibryny przez FDP. Obserwuje się wybroczyny i podbiegnięcia krwawe na błonach śluzowych i skórze, krwawienia z miejsc po wkłuciach dożylnych, krwotoki z dróg rodnych, moczowych i przewodu pokarmowego oraz krwawienia do ośrodkowego układu nerwowego. Z kolei zmiany zakrzepowe w mikrokrążeniu prowadzą do niedokrwiennego uszkodzenia tkanek i narządów. Może rozwinąć się niewydolność nerek, nastąpić upośledzenie wymiany gazowej w płucach, czy dojść do zaburzeń funkcji ośrodkowego układu nerwowego (śpiączka, drgawki). Zakrzepica w drobnych naczyniach skóry objawia się miejscowym zasinieniem, tworzeniem się pęcherzy wypełnionych krwią i martwicą.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Bernard GR, Vincent J-L, Laterre P-F, LaRosa SP, Dhainaut J?F, Lopez-Rodriguez A, Steingrub JS, Garber GE, Heltenbrand JD, Ely W, Fisher CJ for the Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis (PROWESS) Study Group. N Engl J Med 2001; 344: 699-709.
2. Gando S, Kameue T, Nanzaki S, Nakanishi Y: Disseminated intravascular coagulation is a frequent complication of systemic inflammatory syndrome. Thromb Haemost 1996; 75: 224-228.
3. Taylor Jr FB, Toh C-H, Hoots WK, Wada H, Levi M: Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost 2001; 86: 1327-1330.
4. Ten Cate JW, van der Poll T, Levi M, ten Cate H, van Deventer SJH: Cytokines: triggers of clinical thrombotic disease. Thromb Haemost 1997; 78: 415-419.
5. Kopeć M: Udział cytokin w mechanizmach patogene-tycznych rozsianego krzepnięcia śródnaczyniowego. Acta Haemat Pol 1997; 28, Supl 1, 59-67.
6. Mantovani A, Sozzani S, Vecchi A, Introna M, Allavena P: Cytokine activation and endothelial cells: new molecules for an old paradigm. Thromb Haemost 1997; 78: 406-414.
7. Matthay MA: Severe sepsis – a new treatment with both anticoagulant and antiinflamatory properties. N Engl J Med 2001; 344: 759-762.
8. Parrillo JE: Pathogenetic mechanisms of septic shock. N Engl J Med 1993; 328: 1471-1477.
9. Davies MG, Hagen PO: Systemic inflammatory response syndrome. Brit J Surg 1997; 84: 920-935.
10. Stouthard JML, Levi M, Hack CE, Veenhof CHN, Romijn HA, Sauerwein HP, van der Poll T: Interleukin-6 stimulates coagulation, not fibrinolysis, in humans. Thromb Haemost 1996; 76: 738-742.
11. Van der Poll T, Buller HR, ten Cate H, Wortel CH, Bauer KA, van Deventer SJH, Hack E, Sauerwein HP, Rosenberg RD, ten Cate JW: Activation of coagulation after administration of tumor necrosis factor to normal subjects. N Engl J Med 1990; 322: 1622-1627.
12. Esmon CT: Role of coagulation inhibitors in inflammation. Thromb Haemost 2001; 86: 51-56.
13. Faust SN, Levin M, Harrison OB, Goldin RD, Lockhart MS, Kondaveeti S, Laszik Z, Esmon CT, Heyderman RS: Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N Engl J Med 2001; 345: 408-416.
14. Eisele B, Lamy M: Clinical experience with antithrombin III concentrates in critically ill patients with sepsis and multiple organ failure. Semin Thromb Hemost 1998; 24: 71-80.
15. Ten Cate H, Timmerman JJ, Levi M: The patophysiology of disseminated intravascular coagulation. Thromb Haemost 1999; 82: 713-717.
16. Müller-Berghaus G, ten Cate H, Levi M: Disseminated intravascular coagulation: clinical spectrum and established as well as new diagnostic approaches. Thromb Haemost 1999; 82: 706-712.
17. Levi M, de Jonge E, van der Poll T, ten Cate H: Disseminated intravascular coagulation. Thromb Haemost 1999; 82: 695-705.
18. Smith OP, White B: Infectious purpura fulminans: diagnosis and treatment. Brit J Haematol 1999; 104: 202-207.
19. Balk R, Emerson T, Fourrier F, Kruse JA, Mammen EF, Schuster H-P, Vinazzer H: Therapeutic use of antithrombin concentrate in sepsis. Semin Thromb Hemost 1998; 24: 183-194.
20. Fourrier F, Chopin C, Huart J-J, Runge I, Caron C, Goudemand J: Double-blind, placebo-controlled trial of antithrombin III concentrates in septic shock with disseminated intravascular coagulation. Chest 1993; 104: 882-888.
21. Lechner K, Kyrle PA: Antithrombin III concentrates – are they clinically useful? Thromb Haemost 1995; 73: 340-348.
22. Nielsen JD: The effect of antithrombin on the systemic inflammatory response in disseminated intravascular coagulation. Blood Coagul Fibrinolysis 1998; 9 (Suppl 3): 11-15.
23. Vinazzer H: Therapeutic use of antithrombin III in shock and disseminated intravascular coagulation. Semin Thromb Hemost 1989; 15: 347-352.
24. Smith D: 13th Annual Congress of the European Society of Intensive Care Medicine, Rome, Italy, 1-4 October 2000. Crit Care 2000; 4: 347-351.
25. Maruyama I: Recombinant thrombomodulin and activated protein C in the treatment of disseminated intravascular coagulation. Thromb Haemost 1999; 82: 718-721.
26. Bone RC, Balk RA, Cerra FB: Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee. American College of Chest Physicians/Society of Critical Care Medicine. Chest 1992; 101: 1644-1655.
