© Borgis - Postępy Nauk Medycznych 1/2010, s. 86-91
Andrzej Cieśla, *Tomasz Mach
Choroba stłuszczeniowa wątroby w przewlekłym zapaleniu wątroby typu C
Fatty liver disease in chronic hepatitis C
Katedra Gastroenterologii, Hepatologii i Chorób Zakaźnych, Uniwersytet Jagielloński Collegium Medicum w Krakowie
Kierownik Katedry: prof. dr hab. med. Tomasz Mach
Streszczenie
W obrazie histologicznym przewlekłego zapalenia wątroby typu C (pzw C) zmianom zapalno-martwiczym oraz włóknieniu często towarzyszą patomorfologiczne cechy niealkoholowej choroby stłuszczeniowej wątroby (NAFLD). Patogeneza metabolicznych zaburzeń, prowadzących do stłuszczenia z towarzyszącą progresją stanu zapalnego i włóknienia jest złożona i nie w pełni scharakteryzowana. Stłuszczenie w pzw C jest następstwem oddziaływania czynnika wirusowego, szczególnie wyraźnie zaznaczone w przypadku zakażenia gentypem 3 wirusa zapalenia wątroby typu C (HCV), może być także potęgowane czynnikami warunkującymi rozwój NAFLD, stosowaniem leków oraz alkoholem. Zmiany metaboliczne w pzw C pod postacią otyłości trzewnej, zaburzeń metabolizmu glukozy, stłuszczenia wątroby przez niektórych badaczy są określane nazwą „zespołu metaboliczno-wirusowego”. Obecność stłuszczenia zmniejsza skuteczność leczenia przeciwwirusowego interferonem α, przyśpiesza progresję schorzenia do marskości i pierwotnego raka wątroby. Wymagane są dalsze badania mające na celu określenie zależności pomiędzy pzw C i NAFLD oraz ich oddziaływaniem na postęp schorzenia wątroby.
Summary
In the histological examination of chronic hepatitis C (CHC), necroinflammatory changes, and fibrosis are often accompanied by pathomorphological characteristics of non-alcoholic fatty liver disease (NAFLD). Pathogenesis of metabolic disorders leading to steatosis, with the accompanying progression of inflammation and fibrosis, is multifactorial and not fully characterized. Steatosis in CHC is the consequence of the infection of hepatitis C virus (HCV), which is especially noticeable in the case of infection with HCV genotype 3; it can also be intensified by factors determining the development of NAFLD, as well as by drugs or alcohol. Metabolic changes in CHC in the form of visceral obesity, glucose metabolism disorders and fatty liver are defined as the "virology-metabolic syndrome”. The occurrence of steatosis decreases the effectiveness of antiviral treatment with interferon alpha, and induce the progression to cirrhosis and hepatocellular carcinoma. Further studies are needed to determine the relations between CHC and NAFLD and their effects on the progression of liver disease.

Epidemiologia stłuszczenia wątroby w przewlekłym zapaleniu wątroby typu C
Przewlekłe zapalenie wątroby typu C (pzw C) i niealkoholowa choroba stłuszczeniowa wątroby (nonalcoholic fatty liver disease, NAFLD) są najczęstszymi schorzeniami wątroby, mogącymi występować jednocześnie u tych samych pacjentów (1).
NAFLD dotyczy od 20 do 30% populacji krajów Europy i Stanów Zjednoczonych Ameryki Północnej (2). Stłuszczenie stanowi najczęstszą przyczynę przewlekłej choroby wątroby (1). NAFLD jest wątrobową komponentą zespołu metabolicznego zależną od inslinooporności, obecności centralnej otyłości, podwyższonych wartości ciśnienia tętniczego, trójglicerydów, glukozy w surowicy krwi i obniżonego stężenia lipoprotein o wysokiej gęstości (3, 4). W badaniu rezonansem magnetycznym stłuszczenie wątroby obecne jest u 34% populacji dużych miast USA (2). U 33% dawców wątroby stwierdzano stłuszczenie w badaniu histopatologicznym (1). Stłuszczenie wykazuje zróżnicowanie geograficzne zależne od rasy i oddziaływania czynników środowiskowych. We Włoszech, Chinach i Japonii jego obecność dotyczyła odpowiednio 23, 15 i 14% ogólnej populacji (5, 6). NAFLD jest najczęstszą przyczyną podwyższenia AlAT osób poddawanych biopsji wątroby. Epidemia otyłości i cukrzycy typu 2, zwiększa częstość występowania NAFLD (1).
W skali świata zakażonych wirusem zapalenia wątroby typu C (HCV) jest od 100 do 170 milionów osób, co stanowi 1,8-2,3% całej ludzkiej populacji. W częstości występowania stwierdzane są regionalne różnice, najwięcej bo ponad 3% populacji jest zakażonych w Afryce Północnej i Środkowowschodniej, Chinach 2,1%, Europie Wschodniej 2,2%, Stanach Zjednoczonych Ameryki Północnej 1,3%, Azji Południowo-Wschodniej 1,2% i Europie Północnej 1,1% (7, 8).
Statystyczne prawdopodobieństwo jednoczesnego wystąpienia pzw C i NAFLD jest obliczane na 0,36% ogólnej populacji. Rzeczywista częstość równoczesnego wystąpienia obu jednostek chorobowych jest jednak 2,5x większa od oczekiwanej. W przeciwieństwie do autoimmunologicznego zapalenia wątroby (AIH) oraz przewlekłego zapalenia wątroby typu B (pzw B) z około 18% przypadków stłuszczenia, stłuszczenie w przebiegu pzw C jest powszechną cechą tej jednostki chorobowej i występuje od 40 do 86% chorych (9).
Stłuszczenie w przewlekłym zapaleniu wątroby typu C
Stłuszczenie w pzw C może być indukowane zakażeniem HCV lub występować jako zjawisko patogenetycznie tożsame z NAFLD (ryc. 1). Możliwe jest także jednoczesne współistnienie obu powyższych przyczyn (9).
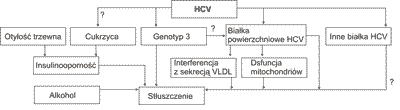
Ryc. 1. Rozwój stłuszczenia wątroby w przewlekłym zapaleniu wątroby typu C (17).
Genotyp 3 wywołuje specyficzną indukcję stłuszczenia miąższu wątroby bez obecności metabolicznych czynników ryzyka stłuszczenia (10). Stłuszczenie w genotypie 3 jest następstwem bezpośredniego steatogennego mechanizmu działania HCV, wykazuje korelację z wielkością wiremii, zanika lub nawraca zależnie od obecności lub braku sukcesu terapeutycznego (1, 10, 11). Powyższych zmian nie obserwuje się w genotypach innych niż 3. W badaniach doświadczalnych wszystkie rodzaje genotypów akumulują śródwątrobowo lipidy, jednak w przypadku genotypu 3 jest ono istotnie większe (12, 13). W genotypie 3 dotyczy 74% przypadków chorych z pzw C w stosunku do 50% zakażonych genotypem innym niż 3 (1).
Współzależność miana HCV RNA genotypu 3 z nasileniem stłuszczenia sugeruje cytopatyczny mechanizm oddziaływania wirusa. Hamowanie tworzenia lipoprotein o bardzo niskiej gęstości (VLDL) z akumulacją śródkomórkową trójglicerydów (TG) jest następstwem zmian błonowych w mitochondriach, uszkadzających oksydację lipidów oraz zahamowania mikrosomalnego transferu TG (14).
Ekspresja białek rdzeniowych HCV in vivo w zakresie miąższu wątroby koreluje z nasileniem stłuszczenia wątroby (14, 15). Ekspresja w zakresie mitochondriów łączy się z hamowaniem α oksydacji kwasów tłuszczowych, syntezą wolnych rodników tlenowych, występowaniem uszkodzeń mitochondrialnego DNA, warunkujących dalsze pogłębianie dysfunkcji mitochondriów (16).
Białka rdzeniowe HCV redukują aktywność mikrosomalnego białka transportującego TG (MTTP). MTTP jest enzymem włączonym w transfer TG, apolipoproteiny B, apolipoproteiny 100 tworzących i wydzielających VLDL (14). Następstwem zahamowania MTTP jest zablokowanie uwalniania VLDL z hepatocytów z równoczesnym śródkomórkowym gromadzeniem lipidów i rozwojem stłuszczenia wątroby (ryc. 2). Obecność w hepatocytach apolipoproteiny A2 wiążącej się z C końcową determinantą białka rdzeniowego HCV umożliwia ochronę MTTP i wydzielanie TG poza komórkę wątrobową (17). Peroksysomowy proliferator α (PPAR α) indukuje ekspresję apolipoprotein A, umożliwiającą pozakomórkową sekrecję apolipoprotein A2 z białkami rdzeniowymi HCV, wznawia sekrecję VLDL i redukuje stłuszczenie (14).
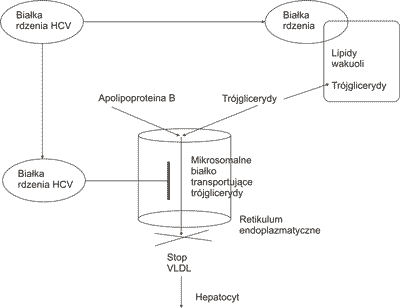
Ryc. 2. Mechanizm stłuszczenia wątroby z udziałem białek wirusa zapalenia wątroby typu C (17).
Pomimo mniej nasilonej insulinooporności (IR), specyficzne dla genotypu 3 HCV jest wątrobowe spichrzanie lipidów w ilościach przewyższających wielkość stwierdzaną w zakażeniu genotypem 1 (10, 18, 19). Część badaczy łączy to zjawisko z obecnością podstawników fenyloalaninowych, charakterystycznych dla białek HCV genotypu 3.
Białka rdzeniowe HCV genotypu 3 zwiększają ekspresję syntazy kwasów tłuszczowych (FAS), głównego enzymu syntezy lipidów (20). Aktywacja promotora FAS jest pośredniczona przez czynnik transkrypcyjny SERB1. Efekt hamowania MTTP jest silniej wyrażony także w przypadku genotypu 3, przebiegającego ze znaczącą redukcją śródkomórkowego stężenia tego białka, w porównaniu z innymi genotypami (21). W genotypie 3 w stosunku do pozostałych widoczne jest silniejsze oddziaływanie wirusa na metabolizm lipidów i wątrobowe stłuszczenie, występujące niezależnie od dodatkowych czynników.
Jeżeli w zakażeniu HCV genotypem 1 stwierdzane jest stłuszczenie to zwykle towarzyszą mu składowe zespołu metabolicznego, w przypadku genotypu 3 zależność ta jest słabiej wyrażona (10, 11). Metaboliczne stłuszczenie wątroby jest współzależne z wielkością wskaźnika masy ciała (BMI), obniża trwałą wirusologiczną odpowiedź, w przypadku leczenia przeciwwirusowego nie znika w następstwie eradykacji HCV (10).
Ze stłuszczeniem w pzw C związana jest przyczynowo otyłość, występująca z częstością od 17 do 38% (10, 22).
IR obserwowana w przypadkach zakażenia HCV wzmacnia lipolizę tkanki tłuszczowej, zwiększa dostępność FFA, w mięśniach szkieletowych promuje niewydajny metabolizm glukozy i w konsekwencji prowadzi do stłuszczenia wątroby (20, 23).
Nie jest jednoznacznie określone czy hiperinsulinizm jest przyczyną, czy następstwem stłuszczenia wątroby. W cukrzycy typu 2 leczenie insuliną chorych ze stłuszczeniem wątroby zmniejsza jego nasilenie, w tym przypadku wydaje się, że hiperinsulinemia jest raczej następstwem, a nie przyczyną stłuszczenia. W stłuszczeniu wątroby możliwy jest zaburzony klirens insuliny warunkujący wzrost jej poziomu we krwi. W tych przypadkach hiperinsulinemia nie jest następstwem komórkowej oporności na insulinę, a wskaźnik HOMA-IR nie jest wiarygodnym wykładnikiem insulinooporności wątroby.
Stan zapalny tkanki tłuszczowej jest przyczyną IR, zwykle łączy się z otyłością. W jego rozwoju duże znaczenie przypisuje się obecności ceramidów powstających z syntezy długołańcuchowych, nasyconych kwasów tłuszczowych. Przeciwnie, obecność w diecie fosfatydylocholiny, krótkołańcuchowych, nienasyconych kwasów zmniejsza aktywność stanu zapalnego. Następstwem stanu zapalnego jest apoptoza adypocytów z mobilizacją makrofagów, powstawaniem nacieków zapalnych, uwalnianiem wolnych rodników tlenowych, TNF α, NFκB, wolnych kwasów tłuszczowych, transkrypcja genów CRP, PAI3. Powyższe czynniki biorą udział w rozwoju stłuszczenia wątroby i zespołu metabolicznego.
Na skutek uwarunkowań cywilizacyjnych i genetycznych pzw C może współistnieć z NAFLD lub NASH. Efekt steatogenny w pzw C może być dodatkowo spotęgowany spożyciem alkoholu, leków, działaniem toksyn.
Histologiczny obraz stłuszczenia w przewlekłym zapaleniu wątroby typu C
Stłuszczenie w badaniu bioptycznym wątroby jest bardzo częstym zjawiskiem w pzw C, w genotypie 3 zmiany są średnio nasilone, w pozostałych przypadkach w większości są łagodne (60-80%), dotyczące mniej niż 30% hepatocytów (24). W zakażeniu HCV dystrybucja tłuszczu jest zwykle ogniskowa, odczyn zapalno-martwiczy i włóknienie lokalizuje się w strefie wrotnej, ciałka Mallory'ego występują rzadziej niż w NAFLD (1). Częstość występowania NASH w populacji ogólnej wynosi 2-3%, w pzw C w zależności od kryteriów rozpoznania histopatologicznego od 5 do 18% (20). Zwykle jest następstwem zakażenia genotypem 3, z towarzyszącym zaawansowanym stłuszczeniem (20). Rozpoznanie NASH w zakażeniu HCV, zwiększa ryzyko szybkiego postępu do marskości oraz wystąpienia HCC (25).
Stłuszczenie i progresja włóknienia w przewlekłym zapaleniu wątroby typu C
Proste stłuszczenie w NAFLD jest rozważane jako łagodne zaburzenie. Stłuszczenie w pzw C zwykle łączy się z obecnością zmian zapalno-martwiczych oraz włóknienia (10, 26). Stłuszczenie może pośredniczyć w wystąpieniu stanu zapalnego wątroby ale także być jego następstwem (26).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Björnsson E, Angulo P: Hepatitis C and steatosis. Arch Med Res 2007; 38(6): 621-7.
2. Browning JD, Szczepaniak LS, Dobbins R et al.: Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004; 40(6): 1387-95.
3. Friis-Liby I, Aldenborg F, Jerlstad P et al.: High prevalence of metabolic complications in patients with non-alcoholic fatty liver disease. Scand J Gastroenterol 2004; 39(9): 864-9.
4. Marchesini G, Brizi M, Bianchi G: Nonalcoholic Fatty Liver Disease. A Feature of the Metabolic Syndrome. Diabetes 2001; 50: 1844-1850.
5. Bedogni G, Miglioli L, Masutti F et al.: Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 2005; 42(1): 44-52.
6. Fan JG, Zhu J, Li XJ et al.: Prevalence of and risk factors for fatty liver in a general population of Shanghai, China. J Hepatol 2005; 43(3): 508-14.
7. Armstrong GL, Wasley A, Simard EP et al.: The prevalence of hepatitis C virus infection in the United States, 1999 through 2002. Ann Intern Med 2006; 144(10): 705-14.
8. Bialek SR, Terrault NA: The changing epidemiology and natural history of hepatitis C virus infection. Clin Liver Dis 2006; 10(4): 697-715.
9. Loguercio C, De Girolamo V, de Sio I et al.: Non-alcoholic fatty liver disease in an area of southern Italy: main clinical, histological, and pathophysiological aspects. J Hepatol 2001; 35(5): 568-74.
10. Adinolfi LE, Gambardella M, Andreana A et al.: Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology 2001; 33(6): 1358-64.
11. Rubbia-Brandt L, Fabris P, Paganin S et al.: Steatosis affects chronic hepatitis C progression in a genotype specific way. Gut 2004; 53(3): 406-12.
12. Abid K, Pazienza V, de Gottardi A et al.: An in vitro model of hepatitis C virus genotype 3a-associated triglycerides accumulation. J Hepatol 2005; 42(5): 744-51.
13. Asselah T, Rubbia-Brandt L, Marcellin P et al.: Steatosis in chronic hepatitis C: why does it really matter? Gut 2006; 55(1): 123-30.
14. Perlemuter G, Davit-Spraul A, Cosson C et al.: Increase in liver antioxidant enzyme activities in non-alcoholic fatty liver disease. Liver Int 2005; 25(5): 946-53.
15. Fujie H, Yotsuyanagi H, Moriya K et al.: Steatosis and intrahepatic hepatitis C virus in chronic hepatitis. J Med Virol 1999; 59(2): 141-5.
16. Ohata K, Hamasaki K, Toriyama K et al.: Hepatic steatosis is a risk factor for hepatocellular carcinoma in patients with chronic hepatitis C virus infection. Cancer 2003; 97(12): 3036-43.
17. Ratziu V, Trabut JB, Poynard T: Fat, diabetes, and liver injury in chronic hepatitis C. Curr Gastroenterol Rep 2004; 6(1): 22-9.
18. Hui JM, Kench J, Farrell GC et al.: Genotype-specific mechanisms for hepatic steatosis in chronic hepatitis C infection. J Gastroenterol Hepatol 2002; 17(8): 873-81.
19. Monto A, Alonzo J, Watson JJ et al.: Steatosis in chronic hepatitis C: relative contributions of obesity, diabetes mellitus, and alcohol. Hepatology 2002; 36(3): 729-36.
20. Blonsky JJ, Harrison SA: Review article: nonalcoholic fatty liver disease and hepatitis C virus – partners in crime. Aliment Pharmacol Ther 2008; 27(10): 855-65.
21. Mirandola S, Realdon S, Iqbal J et al.: Liver microsomal triglyceride transfer protein is involved in hepatitis C liver steatosis. Gastroenterology 2006; 130(6): 1661-9.
22. Hu KQ, Kyulo NL, Esrailian E et al.: Overweight and obesity, hepatic steatosis, and progression of chronic hepatitis C: a retrospective study on a large cohort of patients in the United States. J Hepatol 2004; 40(1): 147-54.
23. Chitturi S, George J: Interaction of iron, insulin resistance, and nonalcoholic steatohepatitis. Curr Gastroenterol Rep 2003; 5(1): 18-25.
24. Lonardo A, Loria P, Adinolfi LE et al.: Hepatitis C and steatosis: a reappraisal. J Viral Hepat 2006; 13(2): 73-80.
25. Ramesh S, Sanyal AJ: Hepatitis C and nonalcoholic fatty liver disease. Semin Liver Dis 2004; 24(4): 399-413.
26. Ghany MG, Kleiner DE, Alter H et al.: Progression of fibrosis in chronic hepatitis C. Gastroenterology 2003; 124(1): 97-104.
27. Leandro G, Mangia A, Hui J et al.: HCV Meta-Analysis (on) Individual Patients' Data Study Group. Relationship between steatosis, inflammation, and fibrosis in chronic hepatitis C: a meta-analysis of individual patient data. Gastroenterology 2006; 130(6): 1636-42.
28. Gabriel A, Ziólkowski A, Radlowski P et al.: Hepatocyte steatosis in HCV patients promotes fibrosis by enhancing TGF-beta liver expression. Hepatol Res 2008; 38(2): 141-6.
29. D'Souza R, Sabin CA, Foster GR: Insulin resistance plays a significant role in liver fibrosis in chronic hepatitis C and in the response to antiviral therapy. Am J Gastroenterol 2005; 100(7): 1509-15.
30. Paradis V, Perlemuter G, Bonvoust F et al.: High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology 2001; 34(4): 738-44.
31. Takuma Y, Nouso K, Makino Y et al.: Hepatic steatosis correlates with the postoperative recurrence of hepatitis C virus-associated hepatocellular carcinoma. Liver Int 2007; 27(5): 620-6.
32. Poynard T, Ratziu V, McHutchison J et al.: Effect of treatment with peginterferon or interferon alfa-2b and ribavirin on steatosis in patients infected with hepatitis C. Hepatology 2003; 38(1): 75-85.
33. Harrison SA, Brunt EM, Qazi RA et al.: Effect of significant histologic steatosis or steatohepatitis on response to antiviral therapy in patients with chronic hepatitis C. Clin Gastroenterol Hepatol 2005; 3(6): 604-9.
34. Bressler BL, Guindi M, Tomlinson G et al.: High body mass index is an independent risk factor for nonresponse to antiviral treatment in chronic hepatitis C. Hepatology 2003; 38(3): 639-44.
35. Lam NP, Pitrak D, Speralakis R et al.: Effect of obesity on pharmacokinetics and biologic effect of interferon-alpha in hepatitis C. Dig Dis Sci 1997; 42(1): 178-85.
