© Borgis - Postępy Nauk Medycznych 11/2011, s. 969-979
*Ewa Brzeziańska, Aleksandra Gładyś, Daria Domańska
Genetic and epigenetic factors in etiopathology of AITD: molecular and clinical implications
Czynniki genetyczne i epigenetyczne w etiopatogenezie AITD: znaczenie molekularne oraz kliniczne
Department of Molecular Bases of Medicine, Medical University of Lodz, Poland
Head of Department: prof. dr hab. med. Ewa Brzeziańska
Streszczenie
W rozwoju autoimmunologicznych chorób tarczycy znaczącą rolę odgrywają interakcje pomiędzy genami, czynnikami środowiskowymi a zmianami epigenetycznymi. W naszej pracy przeglądowej skupiamy się przede wszystkim na czynnikach genetycznych predysponujących do rozwoju choroby Hashimoto oraz choroby Gravesa-Basedowa, zwłaszcza genów zaangażowanych w procesy odpowiedzi immunologicznej: HLA, CTLA-4, CD40, FOXP3, PTPN-22 oraz genów STAT. Epigenetyczna regulacja regionu Xq21-22 oraz Xp11, poprzez metylację promotora może być istotnym mechanizmem zaangażowanym w rozwój AITD. Badania genów immunoregulatorowych udowodniły znaczącą rolę polimorfizmów genów oraz korelacji genotyp-fenotyp w AITD, natomiast regulacja epigenetyczna AITD nie jest dokładnie poznana.
Summary
Autoimmune thyroid diseases (AITDs) are caused by interactions between many genes, environmental factors as well as epigenetic modifications. In present review we focus on genetic background of AITD, mainly on genetic predisposition in hyperthyroid Graves’ disease (GD) and Hashimoto’s (goitrous) thyroiditis, including genes affecting the immune response, mainly genes coding human leukocyte antigens (HLA), cytotoxic T-lymphocyte antigen (CTLA-4), CD40, FOXP3, protein tyrosine phosphatase (PTPN-22), as well as STAT proteins. We point out the possible role of epigenetic regulation mechanism via gene promoter methylation in Xq21-22 and Xp11 regions in AITD development. The importance of studies focused on functional polymorphism variants of immunoregulatory genes, genotype-phenotype correlations, as well as not completely yet recognized epigenetic regulation have also been underlined.

Introduction
Autoimmune thyroid diseases (AITDs), which include hyperthyroid Graves’ disease (GD), Hashimoto’s (goitrous) thyroiditis, atrophic autoimmune hypothyroidism, postpartum thyroiditis (PPT), thyroid-associated orbitopathy (TAO), drug-induced thyroiditis, such as interferon-induced thyroiditis, thyroiditis associated with polyglandural autoimmune syndromes and the presence of thyroid antibodies (Tabs) with no apparent clinical disease, are recognized as multifactorial diseases with vital genetic background. The interaction between environmental and genetic factors cause the impairment of self-tolerance to thyroid autoantigens – cellular and humoral immune responses – directed against the thyroid gland due to AITD developing in genetically predisposed individuals. The twin studies and familial aggregation studies have shown the polygenic basis in Graves’ disease (GD) and Hashimoto’s thyroiditis (HT), the most common endocrine disorders in childhood and adolescence. Both diseases are characterized by the presence of thyroid-reactive T cells and infiltration of the thyroid gland. In GD, the majority of T cells undergo a Th2 differentiation and activate B cells to produce TSH receptor (TSHR) antibodies which stimulate the thyroid resulting in clinical hyperthyroidism. In contrast, HT involves Th1 switching of the thyroid infiltrating T cells which induces apoptosis of thyroid follicular cells and clinical hypothyroidism (1).
In recent years the genetic factors as well as epigenetic background of AITD, particularly GD and HT, have been significantly recognized. Especially, immune regulatory genes affecting the immune response, such as genes for human leukocyte antigens (HLA), cytotoxic T-lymphocyte antigen (CTLA-4), CD40, FOXP3, protein tyrosine phosphatase (PTPN-22), TSHR as well as genes for signal transducer and activator of transcription proteins (STATs) are acknowledged as predisposing to AITD (fig. 1) (2).
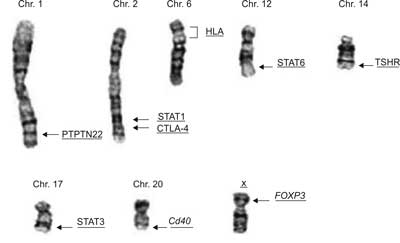
Figure 1. The chromosome localization of main identified AITD susceptibility genes.
Moreover, the epigenetic mechanism is also observed in pathogenesis of autoimmune diseases. The recent study has shown that X chromosome inactivation (XCI) via DNA methylation, may be involved in AITD development, thus explaining the female-predominant tendency (3).
The genetic factors involved in the onset of AITD
Genes for human major histocompatibility complex (MHC)
The MHC region is recognized as a large genomic area that encodes MHC molecules which play an important role in the immune system and autoimmunity. MHC genomic region consists of a complex of genes located on chromosome 6p21. The genes for MHC molecules are divided into 3 regions: (1) Class I genes encode the HLA antigens A, B, and C, (2) Class II genes encode the heterodimeric HLA-DR, DP, and DQ molecules and (3) Class III genes include genes for: complement components (e.g. C4), tumor necrosis factor alpha (TNFα), heat shock protein 70 (Hsp70), and several other genes. The MHC class II molecules are responsible for the initiation of adaptive immune responses. It has been documented that T-cells recognize and respond to antigens when they are attached to the binding groove of an HLA class II molecule (mostly DR and DQ) on the surface of antigen presenting cell. HLA region is highly polymorphic and contains several other immunoregulatory genes, therefore it has been acknowledged as a pivotal candidate locus for AITD as well as for other autoimmune diseases. Additionally, HLA region is recognized as an important chain of immunological synapse involving peptide antigen bound to HLA molecule, T cell receptor, co-stimulatory molecules, receptors on APCs, as well as integrins (4).
Association of HLA with Graves’ disease
Initial studies based on transmission disequilibrium test (TDT) approach, conducted in a large cohort of GD families, have demonstrated strong linkage between GD and HLA. In another studies focused on the role of HMC area in AITD, tough GD association with HLA-DR3(HLA-DRB1*03) has been found. However, in a study from UK only weak evidence for linkage between GD and the HLA region has been recognized, and another study has reported linkage only when conditioning on DR3. Moreover, it has been documented that HLA-B8 and HLA-DQA1*0501 seems to be crucial in GD in Caucasians (5), Additionally, there are reports demonstrating the role of HLA polymorphisms in the clinical manifestation of GD. Interestingly, some studies have documented an association between the likelihood of relapse of GD and HLA-DR3 (5). The increased frequency of HLA-DR3 in patients with Graves’ ophthalmopathy has been observed, but it has not been confirmed by others.
Association of HLA with Hashimoto’s thyroiditis
The association between HLA haplotypes and HT is less definitive than in GD. Early studies failed to find an association between goitrous HT and HLA A- B- or C-antigens. Subsequently, the association of goitrous HT with HLA-DR5 and of atrophic HT with DR3 has been shown (6). The weak association of HT with HLA-DR4 (6) and DR3 (7) in Caucasians was reported in later studies. This observation has been earlier suggested by the studies on animal model in transgenic mice. Finally, HLA-DQW7 (DQB1*0301) was also reported to be associated with HT in Caucasians (8).
Detailed analysis of the HLA class II locus demonstrated that the major HLA haplotype contributing to the shared susceptibility to T1D and AITD was DR3-DQB1*0201, with DR3 conferring most of the shared risk (9).
The effect of ethnicity on the association of HLA with AITD
Many studies have documented that susceptibility of HLA loci to AITD varies among populations, and no consensus has been obtained. In non-Caucasian population HLA alleles were different from those observed in Caucasian groups. Strong association of HLA-DR3 with AITD was observed in Caucasians while in Asian population (for example in Japanese) the associations between HLA-B35 and GD have been found. One study has reported that HLA-DR3, the primary HLA class II allele, predisposes to the joint susceptibility for T1D and AITDs in families in which both diseases cluster. Moreover, a significant association between DRB1*03 and early onset of polyglandular failure has been found, therefore a cooperative susceptibility genes for AITD and other autoimmune diseases have been confirmed (10).
Moreover, an increased frequency of HLA-BW46 has been reported in Chinese population. However, other class I and II HLA alleles have also been reported to be increased in Asian GD patients. The increased frequency of HLA-DRB3*0202 has been observed in African-Americans (11). Interestingly, one study of a mixed population in Brazil demonstrated an association with HLA-DR3, frequent in European group, indicating that this allele may predispose to AITD in different ethnic groups (12). Alternatively, this Brazilian population may have been comprised mostly of European ancestry.
However, two recent genomewide scans in AITD have failed to detect linkage at 6p21 markers (13). On the other hand, whole-genome linkage screening performed by testing a panel of linkage markers of HLA in AITD, has identified the AITD susceptibility gene on: 2q, 6p (HLA), 8q, 10q, 12q,14q and 20q (4).
Reassuming, the available data suggest that HLA involves immune regulatory genes modulating the gene for AITD but not a primary susceptibility gene.
Functional effects of HLA polymorphism
HLA class II molecules are heterodimeric molecules consisting of α and β chain, which form functional highly polymorphic peptide binding pocket (14). In several autoimmune diseases, including AITD, this specific pocket amino acid sequence is recognized as associated with diseases. Moreover, the specific HLA-DR pocket variants have recently been identified as vital for the development of Graves’ disease (GD) (15) and Hashimoto’s thyroiditis (HT).
Sequencing of HLA-DQ genes have documented that arginine at position 74 of the DRβ1 chain is a critical shared amino acid for the development of both GD and HT (15). It has been claimed that a molecular marker of the HLA-DR pocket, determined by specific amino acids, confers a significant risk for the development of AITD as well as T1D – higher than the risk involving the susceptibility genes. The haplotype consisting of HLA-DR pocket amino acids Tyr-26, Leu-67, Lys-71, and Arg-74 has appeared to be strongly associated with AITD and T1D, while amino acids: Leu-26, Phe-26, Ile-67, Asp-70, Glu-71, Ala-71, and Gln-74 have been recognized as protective. Especially, arginine at position 74 of HLA-DRB1 (DRB1-Arg-74), has been shown to be the pivotal in the development of HT and GD (15). Further analysis has shown that the presence of glutamine at position 74 is protective for GD (15). According to functional mechanism of HLA polymorphism, it is hypothesized that the presence of HLA-DR allele with the appropriate amino acids peptide binding pocket, determines the binding of an autoantigenic thyroidal peptide (16). It is recognized that in pocket 4 (P4) of the DR peptide there is a cleft with position 74 of the DRb1chain. Thus, there is a real possibility that an arginine at position 74 changes the structure of the pocket and therefore has an influence on peptide binding and presentation to T-cells. Indeed, structural modeling analysis has shown that the alteration at position 74, from the common neutral amino acids (Ala or Gln) to a positively charged basic amino acid (Arg), significantly converts the three dimensional structure of the P4 peptide binding pocket. This could modify the peptide binding properties of the DR pocket during presentation to T-cells and favor antigenic peptides that induce GD (15). However, this hypothesis awaits confirmation as very few studies have examined the process of binding and presentation of thyroidal autoantigens to T-cells by different HLA-DR subtypes. Interestingly, the results of Hodge et al. (18) study have suggested the interaction at the genetic level between thyroglobulin gene variant and DRB1-Arg74 predisposing to GD. This observation indicates that the thyroglobulin/DRB1-Arg74 genetic interaction is revealed in biochemical relations, in which Arg74 implicates the presentation of thyroglobulin peptides in the initiation phase of GD. Similarly, the recent studies have identified a pocket HLA-DR amino acid signature that presents strong risk for HT.
Cytotoxic T-lymphocyte-associated protein 4 gene (CTLA-4)
The CTLA-4 gene, located on chromosome 2q33, encodes immunoregulatory molecule, expressed on the surface of activated T cells, which via interaction with B7 molecule downregulates immune functions mediated by T-cells activation (19). It is known that T-cells are activated by APCs that present to the T-cell receptor (TCR ) an antigenic peptide bound to an HLA class II protein on the cell surface. However, a second signal is needed for full T-cell activation, including co-stimulatory signals provided by the APCs themselves or other local cells. The CTLA-4 molecule following activation of the TCR is able to transmit signal in response to its ligation with either B7-1 or B7-2 through competition with CD28.
Human CTLA-4 gene is postulated as a major negative regulator of T-cell activity and important genetic factor responsible for susceptibility to variety of autoimmune diseases (20), including: type 1 diabetes mellitus (T1D) (21), asthma (22), Addison’s disease, myasthenia gravis, Sjogren’s syndrome (23), systemic lupus erythematosus (SLE) (24), systemic sclerosis (25), ulcerative colitis (26) and all forms of AITD (GD, HT, as well as the production of thyroid antibodies TAbs) (27).
CTLA-4 gene is highly polymorphic, that has been confirmed in many linkage and association studies in various autoimmune disorders:
1) A to G SNP substitution (49 A/G) at position 49 in exon 1 CTLA-4, resulting in threonine to alanine substitution at codon 17;
2) C to T SNP substitution in the promoter region at position _318 relative to exon 1 start site (_318 C/T);
3) microsatellite polymorphism CTLA-4(AT)n, which is a dinucleotide (AT) repeat in the 3’UTR of the CTLA-4 gene
4) A to G SNP located downstream and outside of the 3’UTR of the CTLA-4 gene (designated CT60).
Recently, it has been claimed that T-effectors activity could be determined by CTLA-4 SNPs. Especially, A49G dimorphism (Thr/Ala exchange in a peptide) leads to the expression of defective receptor, resulting in the inhibitory effect of CTLA-4 molecule on lymphocyte T-cell (28).
The association between AITDs (HT and/or GD) and CTLA-4 polymorphisms (A49G, 1822 C/T and CT60 A/G) and some other polymorphic sites has been confirmed in several studies. Microsatellite polymorphism CTLA-4(AT)n in 3’UTR region has been reported as a first one associated with autoimmune conditions and consistent in different populations (27). In some studies it has been underlined that CTLA-4(AT)n in 3’UTR region is the most powerful polymorphism associated with GD (27). However, particularly CTLA-4 A49G and CT60 polymorphisms have been correlated with susceptibility to AITD development (29). These associations have been consistent irrespective of ethnic backgrounds, and have been found characteristic in many European as well as Asian populations (30).
Functional effects of CTLA-4 polymorphism
Among the all known polymorphism, A/G49 SNP that substitutes threonine for alanine in the signal peptide, leads to misprocessing of CTLA-4 in the ER, resulting in less efficient glycosylation and diminished surface expression of CTLA-4 protein (31). Other reports bring evidence for association between G allele and reduced control of T-cell proliferation (27). It is claimed that this association may be involved in direct effect of the A/G49 SNP or another polymorphism in linkage disequilibrium with the A/G49 SNP. However, the functional studies have revealed that there are no difference in CTLA-4 expression and/or function in case of transiently transfected T-cell line with endogenous CTLA-4 (Jurkat cells), containing a CTLA-4 construct harboring either G or A allele of the A/G49 SNP (32). This observation has suggested that A/G49 is not the causative SNP, but rather remains in linkage disequlibrium with the causative variant.
The results of association studies, focused on C/T_318 SNP of CTLA-4, are also controversial. The study of have confirmed the association of C/T_318 haplotype with CTLA-4 activity, while the results of have been contrary. Recently performed analysis of C/T_318 SNP have established that T allele is related with higher promoter activity in comparison to C allele (33). It has been accepted that the presence of T allele is associated with significantly enhanced expression of CTLA-4 on the surface of stimulated cells, and significantly increased CTLA-4 mRNA level in resting cells (34). Thus, mechanistically, the C/T_318 SNP may influence CTLA-4 levels by changing the binding of a transcription factor LEF-1 (ang. lymphoid enhancing factor 1), via changing TT(C/T)AAG site, which contains the C/T polymorphism (33).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Davies TF. Graves’ diseases: pathogenesis. [In:] Braverman LE, Utiger RD, editors. Werner and Ingbar’s The thyroid: A fundamental and clinical text. Philadelphia: Lippincott Williams & Wilkens 2000; 518-30.
2. Brand OJ, Gough SC. Genetics of thyroid autoimmunity and the role of the TSHR. Mol Cell Endocrinol 2010; 322: 135-43.
3. Yin X, Latif R, Tomer Y et al.: Ann N Y Acad Sci. Thyroid epigenetics: X chromosome inactivation in patients with autoimmune thyroid disease 2007; 1110: 193-200.
4. Tomer Y: Genetic susceptibility to Autoimmune Thyroid Disease: Past, Present and Future. Thyroid 2010; 20: 715-25.
5. Tomer Y, Davies TF.: Searching for the autoimmune thyroid disease susceptibility genes: from gene mapping to gene function. Endocr Rev 2003; 24: 694-717.
6. Petrone A, Giorgi G, Galgani A et al.: CT60 single nucleotide polymorphisms of the cytotoxic T-lymphocyte-associated antigen-4 gene region is associated with Graves’ disease in an Italian population. Thyroid 2005; 15: 232-8.
7. Ban Y, Davies TF, Greenberg DA et al.: The influence of human leucocyte antigen (HLA) genes on autoimmune thyroid disease (AITD): results of studies In HLA-DR3 positive AITD families. Clin Endocrinol (Oxf) 2002; 57: 81-8.
8. Altshuler D, Brooks LD, Chakravarti A: A haplotype map of the human genome. Nature 2005; 437: 1299-320.
9. Golden B, Levin L, Ban Y et al.: Genetic analysis of families with autoimmune diabetes and thyroiditis: evodence for common and unique genes. J Clin Endocrinol Metab 2005; 90: 4904-11
10. Dittmar M, Ide M, Wurm M et al.: Early onset of polyglandular failure is associated with HLA-DRB1*03. Eur J Endocrinol 2008; 159: 55-60.
11. Chen QY, Nadell D, Zhang XY et al.: The human leukocyte antigen HLA DRB3*020/DQA1*0501 haplotype is associated with Graves’ disease in African Americans. J Clin Endocrinol Metab 2000; 85: 1545-9.
12. Maciel LM, Rodrigues SS, Dibbern RS et al.: Association of the HLA-DRB1*0301 and HLADQA1* 0501 alleles with Graves’ disease in a population representing the gene contribution from several ethnic backgrounds. Thyroid 2001; 11: 31-5.
13. Sakai K, Shirasawa S, Ishikawa N et al.: Identification of susceptibility loci for autoimmune thyroid disease to 5q31–q33 and Hashimoto’s thyroiditis to 8q23-q24 by multipoint affected sib-pair linkage analysis in Japanese. Hum Mol Genet 2001; 10: 1379-86.
14. Tomer Y, Menconi F: Type 1 diabetes and autoimmune thyroiditis: the genetic connection. Thyroid 2009; 19: 99-102.
15. Ban Y, Davies TF, Greenberg DA et al.: Arginine at position 74 of the HLA-DRb1 chain is associated with Graves’ disease. Genes Immun 2004; 5: 203-8.
16. Jacobson EM, Huber A, Tomer Y: The HLA gene complex in thyroid autoimmunity: From epidemiology to etiology Journal of Autoimmunity 2008; 30: 58-62.
17. Sawai Y, DeGroot LJ: Binding of human thyrotropin receptor peptides to a Graves’ disease-predisposing human leukocyte antigen class II molecule. J Clin Endocrinol Metab 2000; 85: 1176-9.
18. Hodge SE, Ban Y, Strug LJ et al.: Possible interaction between HLA-DRbeta1 and thyroglobulin variants in Graves’ disease. Thyroid 2006; 16: 351-5.
19. Manzotti CN, Tipping H, Perry LC et al.: Inhibition of human T cell proliferation by CTLA-4 utilizes CD80 and requires CD25+ regulatory T cells. Eur J Immunol 2002; 32: 2888-96.
20. Tomer Y, Greenberg DA, Barbesino G et al.: CTLA-4 and not CD28 is a susceptibility gene for thyroid autoantibody production. J Clin Endocrinol Metab 2001; 86: 1687-93.
21. Kavvoura FK, Ioannidis JP: CTLA-4 gene polymorphisms and susceptibility to type 1 diabetes mellitus: a HuGE Review and meta-analysis. Am J Epidemiol 2005; 162: 3-16.
22. Lee SY, Lee YH, Shin C et al.: Association of asthma severity and bronchial hyperresponsiveness with a polymorphism in the cytotoxic T-lymphocyte antigen-4 gene. Chest 2002; 122: 171-6.
23. Downie-Doyle S, Bayat N, Rischmueller M et al.: Influence of CTLA4 haplotypes on susceptibility and some extraglandular manifestations in primary Sjogren’s syndrome. Arthritis Rheum 2006; 54: 2434-40.
24. Lee YH, Harley JB, Nath SK: CTLA-4 polymorphisms and systemic lupus erythematosus (SLE): a meta-analysis. Hum Genet 2005; 116: 361-7.
25. Almasi S, Erfani N, Mojtahedi Z et al.: Association of CTLA-4 gene promoter polymorphisms with systemic sclerosis in Iranian population. Genes Immun 2006; 7: 401-6.
26. Jiang Y, Xia B, Jiang L et al.: Association of CTLA-4 gene microsatellite polymorphism with ulcerative colitis in Chinese patients. Inflamm Bowel Dis 2006; 12: 369-73.
27. Ban Y, Davies TF, Greenberg DA et al.: Analysis of the CTLA-4, CD28, and inducible costimulator (ICOS) genes in autoimmune thyroid disease. Genes Immun 2003; 4: 586-93.
28. Mäurer M, Loserth S, Kolb-Mäurer A et al.: A polymorphism in the human cytotoxic T-lymphocyte antigen 4 (CTLA4) gene (exon 1 +49) alters T-cell activation. Immunogenetics 2002; 54: 1-8.
29. Takahashi M, Kimura A: HLA and CTLA4 polymorphisms may confer a synergistic risk in the susceptibility to Graves’ disease. J Hum Genet 2010; 55: 323-6.
30. Pastuszak-Lewandoska D, Sewerynek E, Domańska D: CTLA-4 gene polymorphisms and their influence on the predisposition to the autoimmune thyroid diseases (Graves’ disease and Hashimoto’s thyroiditis). Archives Medical of Science 2011; in press.
31. Anjos S, Nguyen A, Ounissi-Benkalha H et al.: A common autoimmunity predisposing signal peptide variant of the cytotoxic T-lymphocyte antigen 4 results in inefficient glycosylation of the susceptibility allele. J Biol Chem 2002; 277: 46478-86.
32. Xu Y, Graves PN, Tomer Y et al.: CTLA-4 and autoimmune thyroid disease: lack of influence of the A49G signal peptide polymorphism on functional recombinant human CTLA-4. Cell Immunol 2002; 215: 133-140.
33. Chistiakov DA, Savost’anov KV, Turakulov RI et al.: Genetic analysis and functional evaluation of the C/T (-318) and A/G(-1661) polymorphisms of the CTLA-4 gene in patients affected with Graves’ disease. Clin Immunol 2006; 118: 233-42.
34. Ligers A, Teleshova N, Masterman T et al.: CTLA-4 gene expression is influenced by promoter and exon 1 polymorphisms. Genes Immun 2001; 2: 145-52.
35. Takara M, Kouki T, DeGroot LJ: CTLA-4 AT-repeat polymorphism reduces the inhibitory function of CTLA-4 in Graves’ disease. Thyroid 2003; 13: 1083-9.
36. Mayans S, Lackovic K, Nyholm C et al.: CT60 genotype does not affect CTLA-4 isoform expression despite association to T1D and AITD in northern Sweden. BMC Med Genet 2007; 8: 3.
37. Yung E, Cheng PS, Fok TF et al.: CTLA-4 gene A-G polymorphism and childhood Graves’ disease. Clin Endocrinol (Oxf) 2002; 56: 649-53.
38. Kinjo Y, Takasu N, Komiya I et al.: Remission of Graves’ hyperthyroidism and A/G polymorphism at position 49 in exon 1 of cytotoxic T lymphocyte-associated molecule-4 gene. J Clin Endocrinol Metab 2002; 87: 2593-6.
39. Allahabadia A, Heward JM, Nithiyananthan R et al.: MHC class II region, CTLA4 gene, and ophthalmopathy in patients with Graves’ disease. Lancet 2001; 358: 984-5.
40. Zaletel K, Krhin B, Gaberscek S et al.: The influence of the exon 1 polymorphism of the cytotoxic T lymphocyte antigen 4 gene on thyroid antibody production in patients with newly diagnosed Graves’ disease. Thyroid 2002; 12: 373-6
41. Zaletel K, Krhin B, Gaberscek S et al.: Thyroid autoantibody production is influenced by exon 1 and promoter CTLA-4 polymorphisms in patients with Hashimoto’s thyroiditis. Int J Immunogenet 2006; 33: 87-91.
42. Van Kooten C, Banchereau J: CD40-CD40 ligand. J Leukoc Biol 2000; 67: 2-17.
43. Bishop GA, Moore CR, Xie P et al.: TRAF proteins in CD40 signaling. Adv Exp Med Biol 2007; 597: 131-51.
44. Tomer Y, Concepcion E, Greenberg DA: A C/T single-nucleotide polymorphism in the region of the CD40 gene is associated with Graves’disease. Thyroid 2002; 12: 1129-35.
45. Kim TY, Park YJ, Hwang JK et al.: A C/T polymorphism in the 50-untranslated region of the CD40 gene is associated with Graves’ disease in Koreans. Thyroid 2003; 13: 919-25.
46. Mukai T, Hiromatsu Y, Fukutani T et al.: A C/T polymorphism in the 50 untranslated region of the CD40 gene is associated with later onset of Graves’ disease in Japanese. Endocr J 2005; 52: 471-7.
47. Heward JM, Simmonds MJ, Carr-Smith J et al.: A single nucleotide polymorphism in the CD40 gene on chromosome 20q (GD-2) provides no evidence for susceptibility to Graves’ disease in UK Caucasians. Clin Endocrinol (Oxf) 2004; 61: 269-72.
48. Jacobson EM, Huber AK, Akeno N et al.: A CD40 Kozak sequence polymorphism and susceptibility to antibody-mediated autoimmune conditions: the role of CD40 tissue-specific expression. Genes Immun 2007; 8: 205-14.
49. Jacobson EM, Concepcion E, Oashi T et al.: A Graves’ disease-associated Kozak sequence single-nucleotide polymorphism enhances the efficiency of CD40 gene translation: a case for translational pathophysiology. Endocrinology 2005; 146: 2684-91.
50. Tuscano JM, Harris GS, Tedder TF: B lymphocytes contribute to autoimmune disease pathogenesis: current trends and clinical implications. Autoimmun Rev 2003; 2: 101-8.
51. Park JH, Chang HS, Park CS et al.: Association analysis of CD40 polymorphisms with asthma and the level of serum total IgE. Am J Respir Crit Care Med 2007; 175: 775-82.
52. Fontenot JD, Gavin MA, Rudensky AY: FOXP3 programs the development and function of CD4ţCD25ţ regulatory T cells. Nature Immunol 2003; 4: 330-6.
53. Holmes D, Knudsen G, Mackey-Cushman S et al.: FoxP3 enhances HIV-1 gene expression by modulating NFkappaB occupancy at the long terminal repeat In human T cells. J Biol Chem 2007; 282: 15973-80.
54. Taylor JC, Gough SC, Hunt PJ et al.: A genome-wide screen in 1119 relative pairs with autoimmune thyroid disease. J Clin Endocrinol Metab 2005; 91: 646-53.
55. Ban Y, Tozaki Tb, Tobe T, Ban Y et al.: The regulatory T cell gene FOXP3 and genetic susceptibility to thyroid autoimmunity: An association analysis in Caucasian and Japanese cohorts Journal of Autoimmunity 2007; 28: 201-7.
56. Ban Y, Tozaki T, Taniyama M et al.: The codon 620 single nucleotide polymorphism of the protein tyrosine phosphatase-22 gene does not contribute to autoimmune thyroid disease susceptibility in the Japanese. Thyroid 2005; 15: 1115-8.
57. Bottini N, Musumeci L, Alonso A et al.: A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 2004; 36: 337-8.
58. Velaga MR, Wilson V, Jennings CE et al.: The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves’ disease. J Clin Endocrinol Metab 2004; 89: 5862-5.
59. Criswell LA, Pfeiffer KA, LumRF et al.: Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620Wallele associates with multiple autoimmune phenotypes. Am J Hum Genet 2005; 76: 561-71.
60. Vang T, Congia M, Macis MD et al.: Autoimmune-associated lymphoid tyrosine phosphatase is a gain-offunction variant. Nat Genet 2005; 37: 1317-9.
61. Ichimura M, Kaku H, Fukutani T et al.: Associations of protein tyrosine phosphatase nonreceptor 22 (PTPN22) gene polymorphisms with susceptibility to Graves’ disease in a Japanese population. Thyroid 2008; 18: 625-30.
62. Dultz G, Matheis N, Dittmar M, et al.: The protein tyrosine phosphatase non-receptor type 22 C1858T polymorphism is a joint susceptibility locus for immunthyroiditis and autoimmune diabetes Thyroid. 2009; 19: 143-8.
63. Chistiakov DA, Savost’anov KV, Turakulov RI et al.: Further studies of genetic susceptibility to Graves’ disease in a Russian population. Med Sci Monit 2002; 8: 180-4.
64. Ho CS, Lam CW, Chan MH, et al.: Electrospray ionisation mass spectrometry: principles and clinical applications. Clin Biochem Rev 2003; 24: 3-12.
65. O J Brand: S C L Gough Genetics of thyroid autoimmunity and the role of the TSHR. Molecular and cellular endocrinology 2010; 322: 135-43.
66. Hodge SE, Ban Y, Strug LJ et al.: Possible interaction between HLA-DRbeta1 and thyroglobulin variants in Graves’ disease. Thyroid 2006; 16: 351-5.
67. Menconi F, Huber A, Osman R et al.: Tg.2098 is a major human thyroglobulin T-cell epitope. J Autoimmun. 2010; 35: 45-51.
68. Stefan M, Jacobson EM, Huber AK et al.: Novel variant of thyroglobulin promoter triggers thyroid autoimmunity through an epigenetic interferon alpha-modulated mechanism. J Biol Chem 2011; 286: 31168-79.
69. Neurath MF, Finotto S: IL-6 signaling in autoimmunity, chronic inflammation and inflammation-associated cancer. Cytokine Growth Factor Rev 2011; 22: 83-9.
70. Marazuela M, García-López MA, Figueroa-Vega N et al.: Regulatory T cells in human autoimmune thyroid disease. J Clin Endocrinol Metab 2006; 91: 3639-46.
71. O’Shea JJ, Murray PJ: Cytokine signaling modules in inflammatory responses. Immunity 2008; 28: 477-87.
72. Trinchieri G review: Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol 2003; 3: 133-46.
73. Imada K, Leonard WJ: The Jak-STAT pathway. Mol Immunol 2000; 37: 1-11.
74. Tamiya T, Kashiwagi I, Takahashi R et al.: Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler Thromb Vasc Biol 2011; 31: 980-5.
75. De Carvalho DD, You JS, Jones PA: DNA methylation and cellular reprogramming. Trends in Cell Biology 2010; 20: 609-17.
76. Becker PB, Hörz W: ATP-dependent nucleosome remodeling. Annu Rev Biochem 2002; 71: 247-73.
77. Xing M: Gene methylation in thyroid tumorigenesis. Endocrinology 2007; 148: 948-53.
78. Fiedler K, Sindrilaru A, Terszowski G et al.: Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood 2011; 117: 1329-39.
79. Thijssen VL, Postel R, Brandwijk RJ et al.: Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc Natl Acad Sci U S A 2006; 103: 15975-80.
80. Juszczynski P, Rodig SJ, Ouyang J et al.: MLL-rearranged B lymphoblastic leukemias selectively express the immunoregulatory carbohydrate-binding protein galectin-1. Clin Cancer Res 2010; 16: 2122-30.
