© Borgis - Postępy Nauk Medycznych 1/2016, s. 31-36
*Małgorzata Sobiecka
Proteinoza pęcherzyków płucnych
Pulmonary alveolar proteinosis
I Klinika Chorób Płuc, Instytut Gruźlicy i Chorób Płuc, Warszawa
Kierownik Kliniki: prof. dr hab. med. Jan Kuś
Streszczenie
Proteinoza pęcherzyków płucnych (ang. pulmonary alveolar proteinosis – PAP), zaliczana do „chorób sierocych”, charakteryzuje się gromadzeniem w pęcherzykach płucnych i końcowych oskrzelach lipidów i białek surfaktantu z upośledzeniem wymiany gazowej. Wyróżnia się trzy postaci PAP: wrodzoną, wtórną i autoimmunologiczną. Najczęściej występuje autoimmunologiczna PAP (90% przypadków), która spowodowana jest obecnością przeciwciał przeciwko czynnikowi stymulującemu kolonie granulocytów/makrofagów (GM-CSF).
Choroba ma zmienny przebieg kliniczny, od spontanicznej remisji zmian do zgonu z powodu niewydolności oddechowej lub nakładającego się zakażenia. Tomografia komputerowa techniką wysokiej rozdzielczości (TKWR) ma istotne znaczenie w postępowaniu diagnostycznym u chorych na PAP, uwidaczniając charakterystyczny dla tej choroby obraz „kostki brukowej” (pogrubienie przegród międzyzrazikowych widoczne na tle obrazu matowej szyby) o geograficznym rozmieszczeniu zmian. Połączenie typowego obrazu klinicznego i radiologicznego w badaniu TKWR z dodatnim wynikiem barwienia płynu z płukania oskrzelowo-pęcherzykowego i wycinka z biopsji przezoskrzelowej płuc pozwala zazwyczaj ustalić pewne rozpoznanie PAP. Dodatkowo wysokie miano autoprzeciwciał w klasie IgG przeciwko GM-CSF jest wysoce czułym i specyficznym markerem choroby. Przez dekady standardowym leczeniem PAP było płukanie całych płuc. Nie wszyscy pacjenci odpowiadają jednak na to leczenie. W autoimmunologicznej postaci PAP zostały zaproponowane nowe metody leczenia, jak podanie GM-CSF podskórnie lub wziewnie, leczenie przeciwciałem anty-CD20 – rituximabem czy plazmaferezą.
Summary
Pulmonary alveolar proteinosis (PAP) is an “orphan lung disorder” characterized by accumulation of surfactant lipids and proteins in the alveoli and terminal airways with resultant impairment in gas exchange. There are three distinct clinical forms: hereditary, secondary and autoimmune. Autoimmune PAP accounts for the vast majority of cases (more than 90%) and is caused by autoantibodies to granulocyte-macrophage colony stimulating factor (GM-CSF).
The condition has a variable clinical course from spontaneous resolution to respiratory failure and death due to disease progression or superimposed infection. The diagnosis of PAP may be strongly supported by high-resolution computed tomography (HRCT) of the chest, which reveals diffuse ground-glass opacification superimposed on septal thickening (“crazy paving”) with geographical distribution. A combination of typical clinical and imaging features with periodic acid-Schiff (PAS)-positive material on bronchoalveolar lavage and transbronchial biopsy is usually sufficient to provide a definitive diagnosis. In addition, a high titer of IgG anti-GM-CSF autoantibodies is highly sensitive and specific for the diagnosis.
For decades, the standard treatment of PAP has been whole lung lavage. However, not all patients respond to this treatment. Thus, in autoimmune PAP new treatment modalities, such as subcutaneous or inhaled GM-CSF, the CD20 antibody – rituximab, and plasmapheresis, have been proposed.

WSTĘP
Proteinoza pęcherzyków płucnych (ang. pulmonary alveolar proteinosis – PAP), po raz pierwszy opisana w 1958 roku przez trzech patologów, jest zaliczana do „chorób sierocych” i charakteryzuje się nieprawidłowym gromadzeniem w świetle pęcherzyków płucnych i dystalnych dróg oddechowych fosfolipidów i białek surfaktantu, co prowadzi do zaburzeń wymiany gazowej, a czasami niewydolności oddechowej (1, 2).
PAP obejmuje niejednorodną pod względem patogenezy, przebiegu klinicznego, rokowania i możliwości leczenia grupę schorzeń. Obecnie uznaje się, że można ją podzielić na dwie kategorie: autoimmunologiczną PAP (dawniej zwaną samoistną), stanowiącą około 90% wszystkich przypadków PAP, i nieautoimmunologiczną PAP, w której wyróżniamy postać wtórną i wrodzoną (3).
Postać wtórna PAP stanowi mniej niż 10% przypadków i rozwija się głównie u dorosłych w przebiegu schorzeń hematologicznych (zespół mielodysplastyczny, białaczka, chłoniak czy szpiczak), przewlekłych zakażeń (Pneumocystis jiroveci lub cytomegalovirusem – CMV), pod wpływem stosowanych leków (chlorambucyl, busulfan, amiodaron, imatinib, leflunomid) lub w wyniku ekspozycji na pyły metali (aluminium, tytan, indium), pyły nieorganiczne (krzemionka, talk, cement) lub organiczne (włókna celulozy, trociny) (2).
Znacznie rzadziej występuje postać wrodzona (ok. 2% przypadków), rozwijająca się u dzieci, spowodowana mutacjami w genach kodujących białka surfaktantu, podjednostki α lub β receptora dla czynnika wzrostu kolonii makrofagów/granulocytów (ang. granulocyte-macrophage colony-stimulating factor – GM-CSF) lub genie kodującym białko transportujące lipidy ABCA3 (2).
Autoimmunologiczna PAP jest najczęstszą postacią choroby, a jej patogeneza wiąże się z obecnością autoprzeciwciał przeciwko cytokinie GM-CSF i tej postaci zostanie poświęcona dalsza część artykułu.
EPIDEMIOLOGIA
Na podstawie obszernych opracowań dużych grup chorych i przekrojowych badań krajowych oszacowano zapadalność na PAP na ok. 0,36-0,49/milion osób, a chorobowość na 3,7-6,2/milion osób w populacji (4, 5).
Pierwsze objawy choroby na ogół występują między 3. a 6. dekadą życia. Dwukrotnie częściej od kobiet chorują mężczyźni, a palacze papierosów (nawet do 79%) oraz osoby narażone na różne pyły stanowią istotny odsetek pacjentów (2).
PATOGENEZA
PAP jest spowodowana nadmiernym gromadzeniem się surfaktantu w świetle pęcherzyków płucnych. Surfaktant, będący mieszaniną fosfolipidów (90%) i towarzyszących białek surfaktantowych (hydrofobowych: SP-B i SP-C oraz hydrofilowych: SP-A i SP-D), syntetyzują i wydzielają pneumocyty typu II. Jego rola w płucach polega na zmniejszaniu napięcia powierzchniowego, co zapobiega zapadaniu się pęcherzyków płucnych i przesiąkaniu osocza z naczyń włosowatych do światła pęcherzyków (2). Surfaktant odgrywa także rolę w odpowiedzi obronnej w płucach poprzez zdolność białek SP-A i SP-D do opsonizacji i bezpośredniego niszczenia patogennych drobnoustrojów. Dzięki zrównoważonej produkcji i usuwaniu surfaktantu utrzymywana jest homeostaza. Pneumocyty typu II i makrofagi pęcherzykowe odgrywają istotną rolę w wychwycie, degradacji, recyklingu i usuwaniu surfaktantu (2). W patogenezie PAP kluczową rolę przypisuje się cytokinie GM-CSF, która jest niezbędna do końcowego dojrzewania makrofagów i usuwania przez nie surfaktantu (3, 6).
Duży postęp w badaniach nad patogenezą PAP dokonał się w 1994 roku, kiedy to przypadkowo odkryto, że u myszy pozbawionych genu kodującego GM-CSF spontanicznie rozwija się schorzenie podobne do PAP (3, 6). Ponadto dowiedziono, że patologiczne zmiany w płucach przypominające PAP można usunąć, przywracając działanie GM-CSF poprzez miejscowe podanie zewnątrzpochodnego GM-CSF, odtworzenie brakującego genu czy przeszczepienie szpiku. W kolejnych latach wykryto w surowicy i w płynie z płukania oskrzelowo-pęcherzykowego (ang. bronchoalveolar lavage – BAL) chorych na tzw. „samoistną” postać PAP przeciwciała neutralizujące przeciwko GM-CSF, które powodowały zaburzenie czynności GM-CSF i jego względny niedobór, prowadząc do zaburzenia funkcji makrofagów pęcherzykowych (2, 3). W postaci wrodzonej i nabytej PAP nie stwierdzono tych przeciwciał. Ponadto w ostatnim czasie wykazano, że przeniesienie wysokooczyszczonych przeciwciał anty-GM-CSF od chorych na „samoistną” postać PAP zdrowym naczelnym odtworzyło cechy patologiczne choroby (3). Wszystkie powyższe dane sugerują, że „samoistna” postać PAP jest nabytą chorobą autoimmunologiczną, w której rozwoju biorą udział przeciwciała skierowane przeciwko GM-CSF.
Obraz kliniczno-radiologiczny
Objawy choroby są niecharakterystyczne. Najczęściej chorzy zgłaszają stopniowo narastającą duszność wysiłkową i przewlekły nieproduktywny kaszel (39-79%), znacznie rzadziej osłabienie, stany podgorączkowe, utratę masy ciała. Przebieg bezobjawowy obserwuje się u ok. 1/3 chorych, a schorzenie zostaje wykryte w oparciu o rutynowo wykonywane badania radiologiczne (4, 5). W badaniu fizykalnym najczęściej stwierdza się trzeszczenia na szczycie wdechu (50%), obecność palców pałeczkowatych (29-40%) i sinicę (20%) (2, 4, 5). Skąpoobjawowy przebieg choroby często prowadzi do kilkumiesięcznego lub kilkuletniego opóźnienia w ustaleniu rozpoznania, jak również zwraca uwagę dysproporcja między nasileniem zmian w badaniach radiologicznych a łagodnymi objawami klinicznymi.
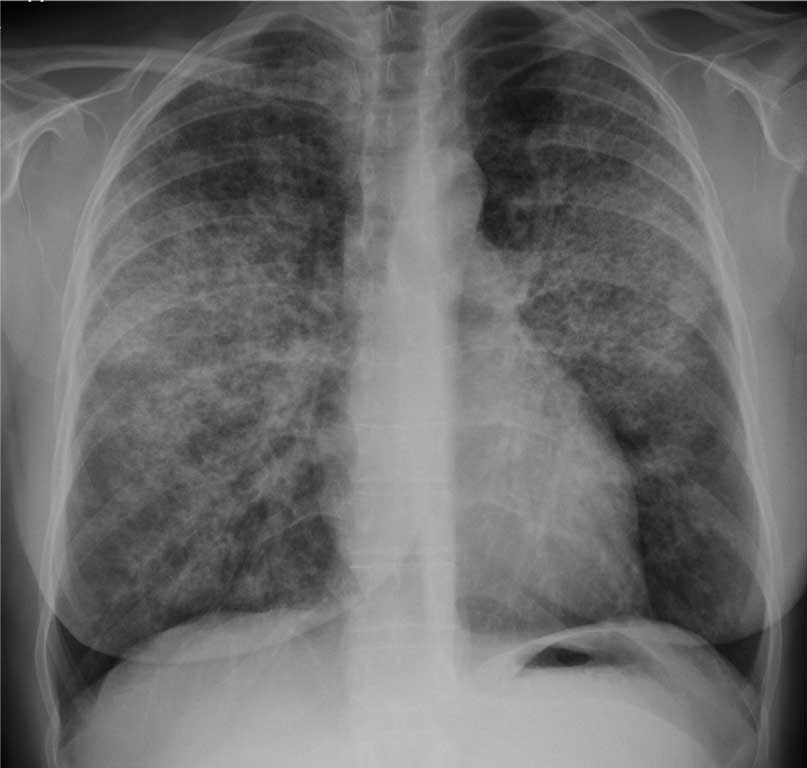
Radiogram klatki piersiowej zazwyczaj pokazuje obustronne, symetryczne zagęszczenia pęcherzykowe, o największym nasileniu zmian w obszarach przywnękowych, co czasami przypomina „obraz skrzydeł motyla” spotykany w obrzęku płuc (ryc. 1). Jednak nie stwierdza się innych radiologicznych cech lewokomorowej niewydolności serca, jak powiększenie sylwetki serca, linie Kerleya B czy obecność płynu w jamach opłucnowych. Rzadziej obserwuje się niesymetryczne lub wręcz jednostronne rozmieszczenie zmian, jak również niejednolite, plamiste rozmieszczenie zmian rozsianych (2, 7).
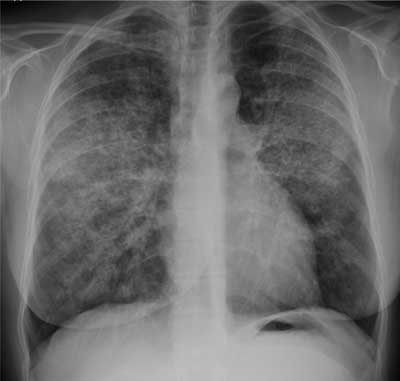
Ryc. 1. Radiogram kobiety chorej na proteinozę pęcherzyków płucnych. Obustronne i symetryczne zagęszczenia pęcherzykowe o największym nasileniu w płatach górnych i środkowych płuc
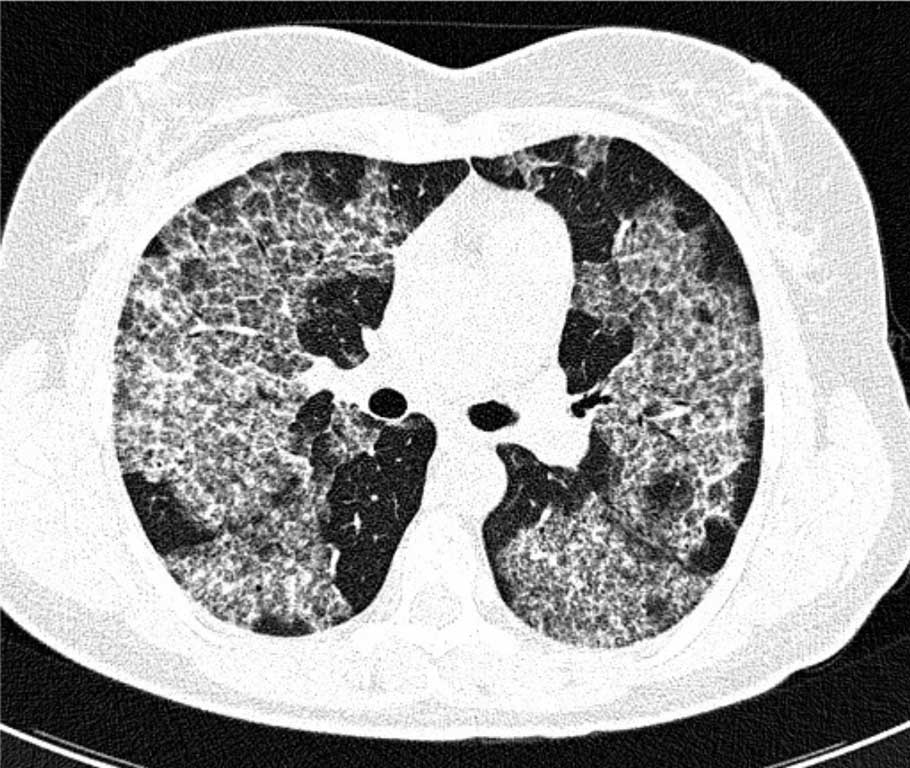
Tomografia komputerowa techniką wysokiej rozdzielczości (TKWR) ma istotne znaczenie w postępowaniu diagnostycznym u chorych na PAP. Obraz zmian jest na tyle charakterystyczny, chociaż nie patognomiczny, że powinien skłonić do oceny płynu z płukania oskrzelowo-pęcherzykowego pod kątem PAP. W badaniu TKWR najczęściej obserwujemy obszary matowej szyby, o geograficznej dystrybucji zmian, obraz zmian siateczkowatych, czasami zagęszczenia miąższowe (na ogół bez bronchogramu powietrznego). Zmiany siateczkowate, imitujące pogrubiałe przegrody międzyzrazikowe, nakładają się na obszary matowej szyby, dając obraz tzw. „kostki brukowej”, charakterystyczny dla PAP (ryc. 2). Podobny obraz „kostki brukowej” może występować w innych schorzeniach, takich jak kardiogenny obrzęk płuc, krwawienie do pęcherzyków płucnych, zakażenia układu oddechowego (pneumocystoza), zewnątrzpochodne lipidowe zapalenie płuc, niektóre podtypy raka płuc (2, 7).
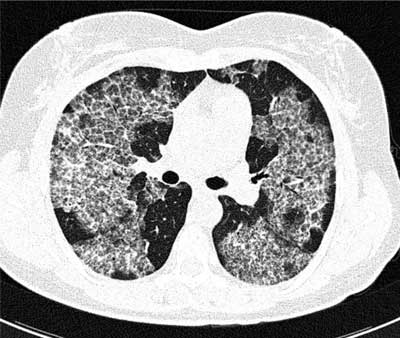
Ryc. 2. TKWR chorej na proteinozę pęcherzyków płucnych. Widoczne zmiany rozsiane o geograficznej dystrybucji pod postacią ognisk matowej szyby zlokalizowanych wśród jednorodnie pogrubiałych przegród tworzących obraz „kostki brukowej”
W badaniach laboratoryjnych najczęściej obserwuje się podwyższone stężenie dehydrogenazy kwasu mlekowego (LDH), antygenu karcinoembrionalnego (CEA), białek surfaktantu SP-A, SP-B i SP-D. Zarówno w surowicy, jak i w BAL-u stwierdza się wyraźny wzrost stężenia mucynopodobnej glikoproteiny KL-6, który koreluje z ciężkością choroby (7).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Rosen SH, Castelman B, Liebow AA et al.: Pulmonary alveolar proteinosis. N Engl J Med 1958; 258: 1123-1142.
2. Wang T, Lazar CA, Fishbein M, Lynch JP: Pulmonary alveolar proteinosis. Semin Respiro Crit Care Med 2012; 33: 498-508.
3. Leth S, Bendstrup E, Vestergaard H, Hilberg O: Autoimmune pulmonary alveolar proteinosis: Treatment options in year 2013. Respirology 2013; 18: 82-91.
4. Seymour JF, Presneill JJ: Pulmonary alveolar proteinosis. Progress in the first 44 years. Am J Respir Crit Care Med 2002; 166: 215-235.
5. Inoue Y, Trapnell BC, Tazawa R et al.: Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med 2008; 177: 752-762.
6. Ben-Dov I, Segel MJ: Autoimmune pulmonary alveolar proteinosis: clinical course and diagnostic criteria. Autoimmunoty Reviews 2014; 13: 13-17.
7. Borie R, Danel C, Debray M-P et al.: Pulmonary alveolar proteinosis. Eur Respir Rev 2011; 20: 98-107.
8. Bonella F, Bauer PC, Griese M et al.: Pulmonary alveolar proteinosis: new insights from a single-center cohort of 70 patients. Respir Med 2011; 105: 1908-1916.
9. Trapnell BC, Uchida K: Pulmonary alveolar proteinosis. Eur Respiro Mon 2009; 46: 208-224.
10. Ramirez-Rivera J, Schulz RB, Dutton RE: Pulmonary alveolar proteinosis: a new technique and rational for treatment. Arch Intern Med 1963; 112: 419-431.
11. Kavuru MS, Malur A, Marshall I et al.: An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur Respiro J 2011; 38: 1361-1367.