Małgorzata Ludzia, Radosław Pietrzak, *Bożena Werner
Zespół Marfana u 7-letniej dziewczynki – opis przypadku
A 7-year-old girl with Marfan syndrome – a case study
Klinika Kardiologii Wieku Dziecięcego i Pediatrii Ogólnej, Warszawski Uniwersytet Medyczny
Kierownik Kliniki: prof. dr hab. n. med. Bożena Werner
Summary
Marfan syndrome is a systemic, autosomal dominant connective tissue disorder with variable expressivity. An early diagnosis is challenging but important, because Marfan syndrome is associated with premature death in untreated patients.
The authors present a case of a 7-year-old girl with Marfan syndrome. The child’s father was diagnosed with Marfan syndrome confirmed by genetic tests. The first symptoms of Marfan syndrome in the presented patient occurred at the age of 2 years, when she presented with mitral and tricuspid valve prolapse, scoliosis, joint hypermobility and body height above 97 percentile. In regular check-ups, aortic root dilatation and the enlargement of the left ventricle were first described one year later. It was decided to introduce beta blocker therapy. Due to the further progression of left ventricular enlargement the girl was given additionally angiotensin II receptor antagonist. In echocardiography follow up no increasing of the aortic root dilatation and the left ventricular enlargement is observed.

Wstęp
Zespół Marfana (ang. Marfan’s syndrome – MFS) jest zaburzeniem tkanki łącznej dotyczącym budowy białka fibryliny, charakteryzującym się zmianami głównie w obrębie układu sercowo-naczyniowego, układu kostno-stawowego oraz narządu wzroku.
Najczęstszymi nieprawidłowościami obserwowanymi w zakresie układu krążenia są: wypadanie płatków, niedomykalność zastawki dwudzielnej, poszerzenie aorty wstępującej, poszerzenie pnia płucnego oraz rozwarstwienie aorty.
Przyczyną nagłego zgonu u pacjentów z zespołem Marfana może być pęknięcie tętniaka aorty, ciężka niedomykalność zastawki mitralnej lub aortalnej. Znaczący wpływ na wydłużanie się w ostatnich latach spodziewanej długości życia u pacjentów z zespołem Marfana ma rozwój technik leczenia kardiochirurgicznego oraz profilaktyczne stosowanie leczenia farmakologicznego: leków beta-adrenolitycznych, inhibitorów konwertazy angiotensyny oraz antagonistów receptora angiotensyny II (1-4).
Opis przypadku
Obecnie 7-letnia dziewczynka od urodzenia pozostaje pod opieką poradni kardiologicznej z powodu wywiadu rodzinnego występowania zespołu Marfana. Ojciec dziecka, u którego rozpoznano zespół Marfana, był dwukrotnie leczony kardiochirurgicznie. W 20. roku życia wszczepiono u niego protezę zastawki aortalnej i aorty wstępującej oraz mechaniczną zastawkę mitralną, natomiast w wieku 36 lat był leczony z powodu rozwarstwienia aorty typu B.
U dziewczynki objawy zespołu Marfana: wysokość ciała > 97 c, wiotkość stawów, wady postawy: asymetryczna budowa klatki piersiowej i skolioza oraz wypadanie płatków zastawki dwudzielnej i trójdzielnej z łagodną niedomykalnością stwierdzono w 2. roku życia. Rodzinne występowanie choroby, mutację w obrębie genu FBN1 potwierdzono badaniem genetycznym.
Początkowo w okresowych kontrolach w poradni kardiologicznej, zarówno w badaniu klinicznym, jak i w badaniach dodatkowych, u dziewczynki nie obserwowano istotnych zaburzeń hemodynamicznych. Według relacji rodziców dobrze tolerowała wysiłek fizyczny, nie zgłaszała żadnych dolegliwości ze strony układu sercowo-naczyniowego. W 3. roku życia po raz pierwszy u dziewczynki zaobserwowano istotne nieprawidłowości w badaniach dodatkowych. W tym czasie w wywiadzie i badaniu przedmiotowym nadal nie obserwowano istotnych objawów klinicznych ze strony układu krążenia poza szmerem skurczowym 3/6 w skali Levine’a z punctum maximum na koniuszku promieniującym wzdłuż lewego brzegu mostka. Parametry życiowe, w tym częstość pracy serca i wartości ciśnienia tętniczego krwi, pozostawały w normie.
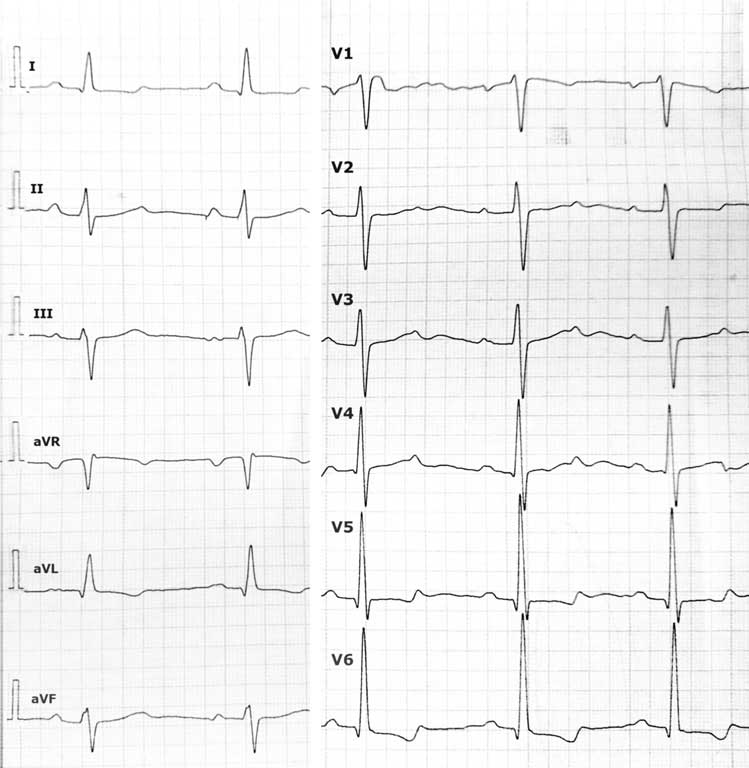
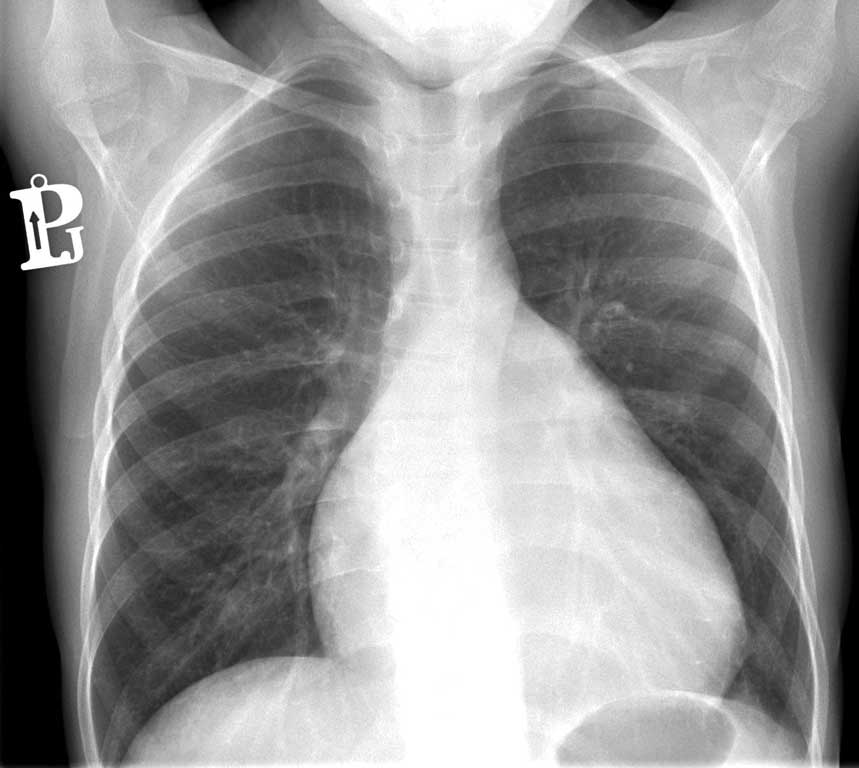
W spoczynkowym 12-odprowadzeniowym zapisie EKG zaobserwowano cechy przerostu i przeciążenia lewej komory (ryc. 1). W badaniu radiologicznym klatki piersiowej stwierdzono powiększoną sylwetkę serca – wskaźnik sercowo-płucny wynosił 0,6 (ryc. 2). W badaniu echokardiograficznym wykazano powiększenie lewej komory (z-score 2,88), wypadanie płatków zastawki mitralnej z umiarkowaną niedomykalnością, niedomykalność zastawki trójdzielnej II stopnia oraz poszerzenie pierścienia zastawki aorty (z-score +3,02) i opuszki (z score +3,53), szerokość aorty wstępującej, jak i w miejscu łącza opuszkowo-tubularnego (ang. sinotubular junction – STJ) były w normie. Zarejestrowano łagodną niedomykalność zastawki aortalnej. Ze względu na obraz echokardiograficzny zdecydowano o włączeniu do leczenia propranololu.
Ryc. 1. 12-odprowadzeniowe EKG u dziewczynki
Ryc. 2. Zdjęcie radiologiczne klatki piersiowej u dziewczynki (wskaźnik sercowo-płucny 0,6)
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Pereira L, Levran O, Ramirez F et al.: A molecular approach to the stratification of cardiovascular risk in families with Marfan’s syndrome. N Engl J Med 1994; 21: 191-193.
2. Hoffjan S: Genetic dissection of Marfan syndrome and related connective tissue disorders: an update 2012. Mol Syndromol 2012; 3: 47-58.
3. Canadas V, Vilacosta I, Bruna I, Fuster V: Marfan syndrome, part 1: pathophysiology and diagnosis. Nat Rev Cardiol 2010; 7: 256-265.
4. Judge D, Dietz H: The Marfan’s syndrome. Lancet 2005; 366: 1965-1976.
5. Loeys BL, Dietz HC, Braverman AC: The revised Ghent nosology for the Marfan syndrome. J Med Genet 2010; 47: 476-485.
6. Von Kodolitsch Y, Robinson P: Marfan syndrome: an update of genetics, medical and surgical management. Heart 2007; 93(6): 755-760.
7. Castellano J, Silvay G, Castillo J: Marfan Syndrome. Clinical, Surgical, and Anesthetic Considerations. Semin Cardiothorac Vasc Anesth 2014; 18(3): 260-271.
8. Shores J, Berger KR, Murphy EA, Pyeritz RE: Progression of aortic dilatation and the benefit of long term beta-adrenergic blockade in Marfan’s syndrome. N Engl J Med 1994; 330(19): 1335-1341.
9. Keane MG, Pyeritz RE: Medical management of Marfan syndrome. Circulation 2008; 117: 2802-2813.
10. Canadas V, Vilacosta I, Bruna I, Fuster V: Marfan syndrome, part 2: treatment and management of patients. Nat Rev Cardiol 2010; 7: 266-276.
11. Ladouceur M, Fermanian C, Lupoglazoff JM et al.: Effect of beta-blockade on ascending aortic dilatation in children with Marfan syndrome. Am J Cardiol 2007; 99(3): 406-409.
12. Habashi J, Judge D, Holm T et al.: Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science 2006; 312(5770): 117-121.
13. Groenink M, den Hartog AW, Franken R et al.: Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J 2013; 34(45): 3491-3500.
14. Forteza A, Evangelista A, Sánchez V et al.: Efficacy of losartan vs. atenolol for the prevention of aortic dilation in Marfan syndrome: a randomized clinical trial. Eur Heart J 2016; 37(12): 978-985.
15. Franken R, den Hartog AW, Radonic T et al.: Beneficial Outcome of Losartan Therapy Depends on Type of FBN1 Mutation in Marfan Syndrome. Circulation 2015; 8(2): 383-388.
16. Muiño-Mosquera L, De Nobele S, Devos D et al.: Efficacy of losartan as add-on therapy to prevent aortic growth and ventricular dysfunction in patients with Marfan syndrome: a randomized, double-blind clinical trial. Acta Cardiol 2017; 72(6): 616-624.
17. McLoughlin D, McGuinness J, Byrne J et al.: Pravastatin reduces Marfan aortic dilation. Circulation 2011; 124: S168-173.
18. Stein LH, Berger J, Tranquilli M et al.: Effect of statin drugs on thoracic aortic aneurysms. Am J Cardiol 2013; 112: 1240-1245.
19. Jost CHA, Greutmann M, Connolly H et al.: Medical Treatment of Aortic Aneurysms in Marfan Syndrome and other Heritable Conditions. Curr Cardiol Rev 2014; 10(2): 161-171.
20. Clarice Yang HH, Jong Moo K, Chum E et al.: Effectiveness of combination of losartan potassium and doxycycline versus single-drug treatments in the secondary prevention of thoracic aortic aneurysm in Marfan syndrome. J Thorac Cardiovasc Surg 2010; 140(2): 305-312.