© Borgis - Nowa Pediatria 2/2002, s. 58-62
Ivo Marik1, Kazimierz Kozlowski2
Dysplazja czołowo-przynasadowa (FMD) u 3,5-letniej dziewczynki i jej „zdrowej” matki
Frontometaphyseal dysplasia (FMD) in a 3 and 5/12 year old girl and in the „unaffected” mother
1Ambulant Centre for Defects of Locomotor Apparatus, Prague, Czech Republic
2Royal Alexandra Hospital for Children, Sydney, Australia
Streszczenie
We report a girl with frontometaphyseal dysplasia in whom the first phenotypic abnormality noted at the age of 3 months was metopic suture torus. At the end of the first year of life knock knee deformity and torsion of the legs was noted and this progressed in spite of orthotic treatment. The diagnosis of frontometaphyseal dysplasia was established at the age of 3 years and 5 months. Family studies revealed that the mother, supposed to be normal was also affected. Previously unreported clinical manifestations and radiographic findings are documented to further delineate and expand the spectrum of the disorder.

WSTĘP
Dysplazja czołowo-przynasadowa (frontometaphyseal dysplasia, FMD) jest rzadkim schorzeniem rozpoznawanym zwykle u dzieci w wieku szkolnym. Przedstawiamy opis przypadku choroby dziewczynki, u której pierwszą stwierdzaną anomalią był kostny wał nadoczodołowy w obrębie szwu czołowego stwierdzony w 3 miesiącu życia. Pod koniec pierwszego roku życia zauważono zmiany w obrębie stawu kolanowego i koślawość kolan. U matki dziewczynki cechy tego zespołu były miernie nasilone. Uważano ją za zdrową osobę aż do czasu ustalenia rozpoznania u córki (ryc. 1a).

Ryc. 1a. Wygląd twarzy dziewczynki w wieku 4,5 roku oraz matki w wieku 28 lat. Zwraca uwagę wybitne zgrubienie łuków brwiowych, zarówno u matki, jak i córki, oraz wał kostny w obrębie szwu czołowego u córki.
OPISY PRZYPADKÓW
Przedstawiana przez nas dziewczynka czeskiego pochodzenia urodziła się z ciąży 1, porodu 1, w 36 tygod-niu ciąży. Matka w chwili porodu miała 24 lata. Rodzice dziecka byli zdrowi. Ciąża powikłana była zakrzepicą żył lewej kończyny dolnej, matka leczona była drobnocząsteczkową heparyną. Po urodzeniu dziewczynka ważyła 2800 g i miała 49 cm długości, wymagała tlenoterapii, żywienia pozajelitowego i leczenia antybiotykami przez 5 pierwszych dni życia z powodu anemii i podejrzenia infekcji wewnątrzmacicznej. W badaniu ultrasonograficznym mózgu stwierdzono wylew podwyściółkowy I stopnia, a w badaniu echo serca ubytek międzyprzedsionkowy, który uległ samoistnemu zamknięciu. W wieku 3 miesięcy zauważono u niej kostny wał wzdłuż szwu czołowego, a pod koniec pierwszego roku życia postępujące zniekształcenia dotyczące stawów kolanowych i kończyn dolnych. Zmiany w obrębie kończyn dolnych nasilały się w ciągu kolejnych lat obserwacji mimo leczenia ortopedycznego. Rozwój psychoruchowy dziewczynki w okresie obserwacji był prawidłowy. W wieku 3 lat, w trakcie badań w oddziale genetyki w Ostrawie, wysunięto u dziewczynki podejrzenie dysplazji przynasadowej.
W wieku 3,5 roku dziewczynka była badana w Ośrodku dla Pacjentów z Chorobami Narządu Ruchu w Pradze. Stwierdzono wysokość ciała 103 cm (75 centyl), masa ciała 17 kg (90 centyl), obwód głowy 50 cm (75 centyl) i obwód klatki piersiowej 57 cm (65 centyl). Stwierdzono także obustronną koślawość stawów kolanowych z łukowatym wygięciem kończyn dolnych, bardziej nasiloną po stronie lewej (ryc. 1b). Zakres ruchów w stawach biodrowych oraz ruchów zginania w stawach kolanowych był zmniejszony. Kończyny były zbyt długie w stosunku do tułowia. Dodatkowo, na skutek asymetrii długości kończyn dolnych doszło do skrzywienia kręgosłupa. Stwierdzono nietypowy wygląd twarzy; wysokie czoło z charakterystycznym wałem kostnym wzdłuż szwu czołowego oraz przerost części kostnych okolicy nadoczodołowej. Dziewczynka miała niewielki hiperteloryzm, wytrzeszcz gałek ocznych, obustronnie zmarszczkę nakątną, długą rynienkę podnosową i niedorozwój żuchwy. Zęby były hipoplastyczne, pomiędzy siekaczami widoczny rozstęp. Miała długie, proste, brązowe włosy. W wieku 6 lat ważyła 26 kg (90 centyl), miała wzrost 119,7 cm (50-75 centyl). Badanie proporcji ciała wykazało nadmiernie długie kończyny górne (+1,5 SD) i dolne (+2 SD), krótką szyję (-1,2 SD), stosunkowo wąskie ramiona (-0,6 SD). Miednica i klatka piersiowa były poszerzone (odpowiednio: +2,5 SD i +1,5 SD). Czaszka była wydłużona w kierunku przednio-tylnym (łódkogłowie). Obwód głowy wynosił 52,3 cm (75-90 centyl). Rozwój psychoruchowy dziewczynki był prawidłowy. Nie stwierdzono upośledzenia słuchu. Poszerzone badania laboratoryjne, zwłaszcza dotyczące gospodarki wapniowo-fosforanowej, nie wykazały nieprawidłowości.
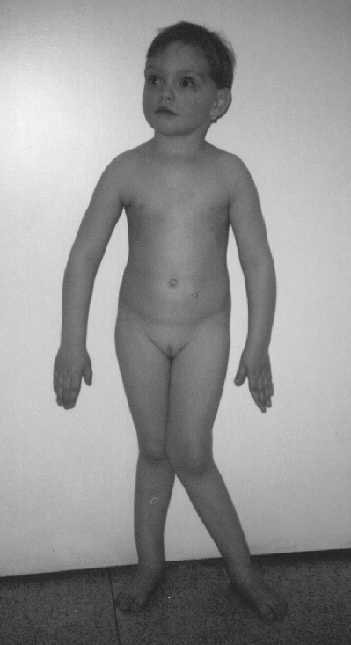
Ryc. 1b. Dziewczynka w wieku 3,5 roku. Nasilona koślawość lewego stawu biodrowego. Miernie nasilona koślawość stawu biodrowego prawego i boczne wygięcie prawej kończyny dolnej.
Badania radiologiczne ujawniły typowe dla dysplazji czołowo-przynasadowej zmiany: zwiększoną gęstość kości w obrębie podstawy czaszki i środkowej części kości czołowej, brak zatok czołowych. Zęby były hipoplastyczne/dysplastyczne. Żebra miały nieregularne zarysy, były cienkie (ryc. 2a). Łopatki duże, dysplastyczne z wydatnymi wyrostkami barkowymi, a talerze kości biodrowych małe i wachlarzowato poszerzone. Stwierdzono koślawość stawów biodrowych (ryc. 2b). Wszystkie kości długie były nieznacznie łukowato wygięte, a ich przynasady wykazywały niedostateczne modelowanie. Najbardziej zniekształconymi kośćmi były piszczele, które w projekcji bocznej przybierały kształt litery „S”. Rzepki były małe (ryc. 2c), obraz kręgosłupa prawidłowy. W obrębie kości dłoni nie stwierdzono znaczących zaburzeń (ryc. 2d).
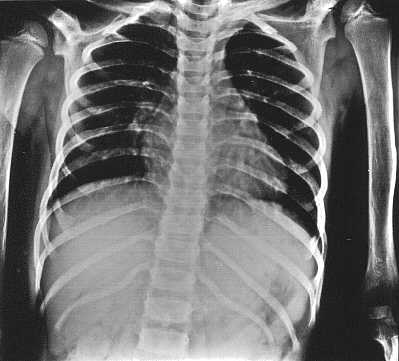
Ryc. 2a. Obraz klatki piersiowej dziewczynki w wieku 5 lat i 10/12. Cienkie żebra. Obustronne, łukowate zniekształcenie 12 żebra.
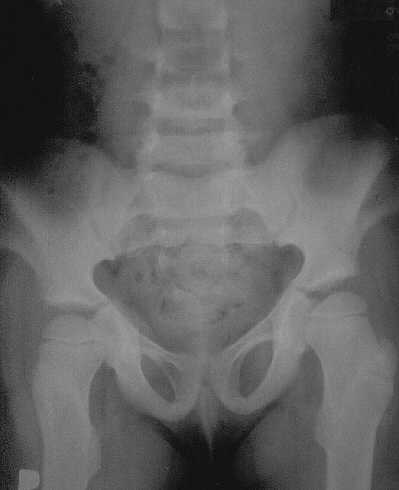
Ryc. 2b. Kości miednicy u dziewczynki w wieku 6 lat. Wachlarzowate poszerzenie talerzy kości biodrowych. Obustronna koślawość w stawach biodrowych.
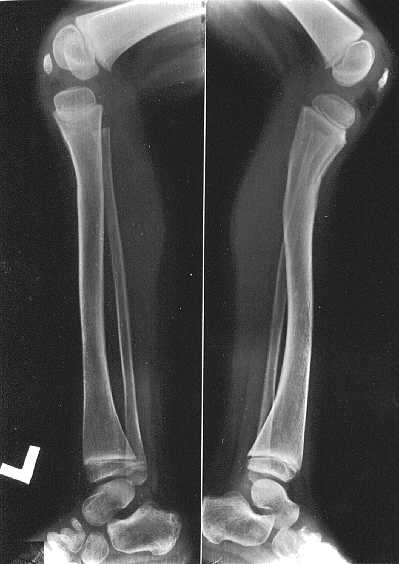
Ryc. 2c. Obraz podudzi u dziewczynki w wieku 4 lat i 10/12. Obustronne wygięcie piszczeli w kształcie litery "S", bardziej nasilone po stronie prawej, małe rzepki.
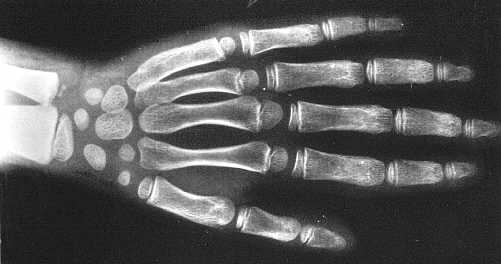
Ryc. 2d. Obraz dłoni dziewczynki w wieku 6 lat. Poszerzenie i niedostateczne modelowanie paliczków, szczególnie dotyczące paliczków środkowych. Niewielkie zmiany dysplastyczne kości nadgarstka i śródręcza. Wiek kostny odpowiada wiekowi kalendarzowemu.
Matka opisywanej dziewczynki miała wygląd twarzy podobny do córki. Jej wzrost wynosił 162 cm, masa ciała 56 kg. Stwierdzano u niej niewielką koślawość kolan ale bez wygięcia kończyn dolnych, miała prawidłowy chód. Badania radiologiczne miednicy, czaszki i lewej dłoni, ujawniły u niej charakterystyczne cechy dysplazji czołowo-przynasadowej (ryc. 3a-c).
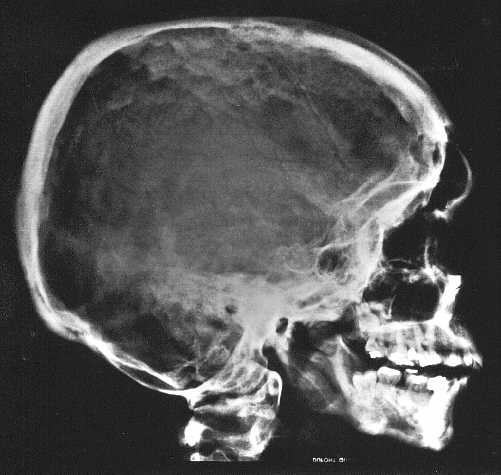
Ryc. 3a. Zwiększona gęstość podstawy i sklepienia czaszki oraz kości twarzoczaszki, sklerotyzacja szwów czaszkowych, nieprawidłowe ustawienie stawu szczytowo-potylicznego, niedorozwój zatoki czołowej, słabe upowietrzenie wyrostków sutkowatych, wydatne brzegi kostne nadoczodołowe, hipoplazja szczęki i żuchwy, niedorozwój zębów.
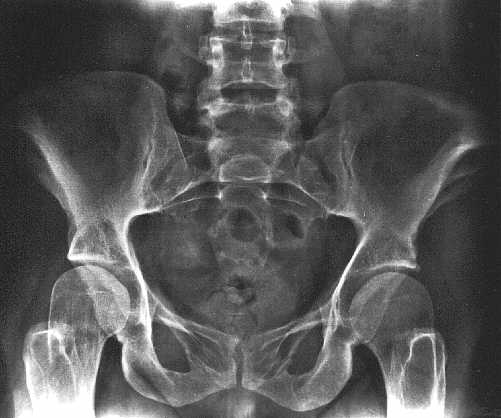
Ryc. 3b. Małe, wachlarzowato poszerzone talerze kości biodrowych, koślawość stawów biodrowych, poszerzone szyjki kości udowych.
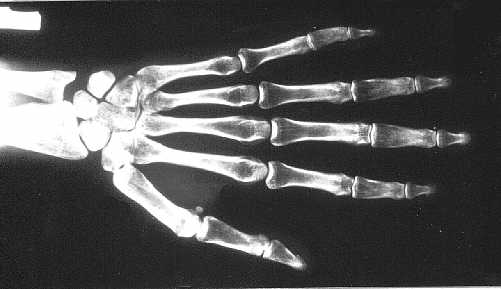
Ryc. 3c. Poszerzenie i niedostateczne modelowanie środkowych paliczków dłoni.
DYSKUSJA
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
24 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
59 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
119 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 28 zł
Piśmiennictwo
1. Gorlin R.J., Cohen M.M.: Frontometaphyseal dysplasia. A new syndrome. Am. J. Dis. Child 1969, 118:487-494. 2. Taybi H., Lachmann R.S.: Radiology of syndromes, metabolic disorders and skeletal dyslasias 4th edn. Year Book, Chicago 1996, 821:23. 3. Holt J.F. et al.: Frontometaphyseal dysplasia. Radiol. Clin. North Am. 1972, 10:225-243. 4. Danks D.M. et al.: Frontometaphyseal diysplasia. Am. J. Dis. Child. 1972, 123:254-258. 5. Kassner E.G. et al.: Frontometaphyseal dysplasia: evidence for autosomal dominat inheritance. Am. J. Roentgenol 1976, 127:927-933. 6. Weiss L. et al.: Frontometaphyseal dysplasia. Evidence for dominant inheritance. Am. J. Dis. Child. 1976, 130:259-261. 7. Medlar R.C., Crawford A.H.: Frontometaphyseal dysplasia. J. Bone Joint Surg. (Am) 1978, 60A:392-394. 8. Kanemura T. et al.: Frontometaphyseal dysplasia: Evidence for autosomal dominant inheritance. Am. J. Roentgenol 1979, 127:927-933. 9. Gorlin R.J., Winter R.B.: Frontometaphysela dysplasia – Evidence for X-linked inheritance. Am. J. Med. Genet. 1980, 5:81-84. 10. Fitzsimmons J.S. et al.: Frontometaphyseal dysplasia. Further delineation of the clinical syndrome. Clin. Genet. 1982, 22:195-205. 11. Park J.M. et al.: Mitral valve prolaps in a patient with frontometaphyseal dysplasia. Clin. Pediatr (Phila) 1986, 25:469-471. 12. Spranger J.W. et al.: Bone dysplasias. Saunders Co. Philadelphia 1974, 321-322. 13. Beighton P., Cremin B.J.: Sclerosing Bone Dysplasias. Springer. Berlin 1980, 75-81. 14. Bieganski T., Makowski A.: Dysplazja czołowo-przynasadowa. Przegląd Radiol. 1995, 60:100-102. 15. Nishimura G. et al.: Radiological changes of frontometaphyseal dysplasia in the neonate. Ped. Radiol. 1995, 25:143-146.