© Borgis - Postępy Nauk Medycznych 7/2010, s. 553-561
*Andrzej Pławski1, Marta Podralska1, Piotr Krokowicz3, Jacek Paszkowski2, Ryszard Słomski1
Familial adenomatous polyposis of colon
Rodzinna polipowatość gruczolakowata jelita grubego
1Institute of Human Genetics, Polish Academy of Sciences
Head of Institute of Human Genetics: prof. dr hab. Jerzy Nowak
2Department of General and Colorectal Surgery, Karol Marcinkowski University of Medical Sciences
Head of Department of General and Colorectal Surgery: prof. UM dr hab. med. Piotr Krokowicz
3Department of General, Gastroenterological and Endocrine Surgery, Karol Marcinkowski Medical University
Head of Department of General, Gastroenterological and Endocrine Surgery: prof. dr hab. med. Michał Drews
Streszczenie
Rodzinna polipowatość jelita (FAP) jest dobrze poznanym zespołem predyspozycji do występowania choroby nowotworowej, dziedzicznym w sposób autosomalno dominujący ze skłonnością do występowania dużej liczby polipów w okrężnicy i odbytnicy. Pierwszymi objawami FAP są biegunka i krew w stolcu. Wraz z rozwojem choroby nowotworowej występuje utrata masy ciała i osłabienie. FAP występuje z częstością 1 na 10 000 nowourodzonych. Czas wystąpienia polipów i ich liczba są w przypadku tej choroby zróżnicowane. Klasyczna forma FAP charakteryzuje się występowaniem więcej niż 100 polipów, które pojawiają się w drugiej dekadzie życia. Średni wiek występowania polipów to 15 lat. Najmłodszy chory miał 3 lata. Polipy charakteryzują się dużym potencjałem rozwoju w kierunku guza złośliwego. Rozwój nowotworu złośliwego może następować od późnego dzieciństwa do 70. roku życia. Łagodna forma polipowatości rodzinnej charakteryzują się łagodniejszym przebiegiem niż klasyczny FAP. Występowanie FAP jest związane z mutacjami w genie supresorowym nowotworów APC, który został opisany w 1991 r. Gen APC jest zlokalizowany na chromosomie 5q21 i jest związany z kontrolą proliferacji komórek. Forma recesywna FAP jest spowodowana przez homozygotyczne mutacje w genie MUTYH. Gen MUTYH jest zaangażowany w naprawę z oksydacyjnych uszkodzeń DNA. Polipowatość związana z MUTYH (MAP) jest skłonnością do występowania polipów jelita, ale liczba polipów jest niższa w klasycznym FAP. Wysokie ryzyko wystąpienia choroby nowotworowej w tych chorobach sprawia, że należą do ważnych zagadnień medycznych. W Polsce badania molekularne wykonywane są od ponad dziesięciu lat. W Instytucie Genetyki Człowieka PAN w Poznaniu utworzono Bank DNA dla polskich pacjentów FAP. W Banku DNA zgromadzono próbki DNA od czterystu rodzin z FAP.
Summary
Familial adenomatous polyposis (FAP) is a well-known predisposition for the occurrence of a large number of polyps in the colon and rectum inherited in an autosomal dominant manner. First symptoms of FAP are diarrhea and blood in stool. Weight loss and weaknesses occur after the development of advanced tumour. The incidence of the FAP disorder is one per 10 000 newborns. There is a high heterogeneity with regard to the number and time of the occurrence of polyps. The classical form of FAP is characterized by the occurrence of more than 100 polyps which appear in the second decade of life. The average time of occurrence of polyps is 15 years. The earliest symptoms of polyposis were observed in a three-year-old child. The polyps are characterized by large potential for the development towards malignant tumour. Turning into malignancy can occur from late childhood to the age of 70. Attenuated adenomatous polyposis coli cases are characterized by benign course of disease as opposed to the classical FAP. The occurrence of FAP is associated with mutations in the APC tumour suppressor gene, which was described in 1991. The APC gene is a tumour suppressor gene located on chromosome 5q21 involved in cell proliferation control. The recessive form of FAP is caused by mutation in the MUTYH gene occurring in homozygotic state. The MUTYH gene is involved in a repairing of the oxidative DNA damage. MUTYH associated polyposis (MAP) is predisposition for the occurrence of polyps of the colon but the number of polyps is lower in comparison to classical FAP. The high risks of cancer observed in those syndromes make them important medical issues. Molecular studies of colon polyposis have been performed in Poland for over ten years. A DNA Bank for Polish FAP patients was established at the Institute of Human Genetics in Poznań in which DNA samples from four hundred FAP families were collected.

INTRODUCTION
Familial adenomatous polyposis (FAP) constitutes approximately 1% of all large intestine tumour cases (1). The disease occurs de novo with the frequency of 1 for 8000 to 10 000 births (2) and the age of symptom appearance in patients varies quite considerably and differences in symptoms appearance are also recorded between siblings. However, it can be assumed that the occurrence of the large intestine tumour in young age should give a signal to perform a family anamnesis which makes it possible to determine if high hereditary predisposition for the disease occurs in the family (3). Occurrence of a single case of the disease does not rule out a high hereditary predisposition because a given patient may be the first carrier of the mutation. FAP symptoms occur earlier than in HNPCC and appear in the second decade of life, although cases of the disease occurrence were reported as early as at the age of 5 and in the group of our youngest patients, one patient was only 3 when polyps were diagnosed (4). The genetic background of the appearance of adenomatous polyps is the occurrence of mutations in APC genes in the case of FAP and in MUTYH – in the case of the recessive form of adenomatous polyposis.
The APC gene is a tumour suppressor gene. Tumour suppressor genes are involved in cell proliferation control. Protein products of suppressor genes participate in the control of the cell cycle as its inhibitors and are also constituents of the growth inhibition contact system. Suppressor genes are involved in the mechanisms of maintaining cell numbers at a constant level and disturbances in those instruments result in the frequency increase of cell divisions as well as in greater numbers of errors developing in the course of divisions. This leads to the accumulation of changes in genetic material and selection of an immortal clone of very frequent cell divisions capable of settling other tissues.
In the case of suppressor genes, mutation phenotype is masked by a correct gene allele. There are two independent mutations within the suppressor gene locus during the initiation of the neoplastic process (5). In the case of carrier state of the mutated allele of the APC gene, the risk of occurrence of the second mutation, consequently, of the initiation of the neoplastic disease is very high.
History of investigations of the APC gene
FAP was identified as a heritable pathogenic syndrome already in 1920s. In 1972, Gardener syndrome was described which is a form of FAP characterised not only by the presence of hundreds or even thousands of polyps in the intestine but also of osteomas and retinal hyperpigmentation ( congenital hypertrophy of the retinal pigment epithelium – CHRPE). The occurrence of FAP was associated with the q21-q22 region of chromosome 5 on the basis of an extensive deletion discovered in cytological investigations as well as research results of RFLP marker association in a patient with the Gardener syndrome and advanced polyp growth in the colon (6). Towards the end of 1980s, association studies directed the quest for the gene towards the region which encompassed APC and MCC genes separated from each other by 150 bp. In 1991, the following three genes: DP1, SRP19 and DP2.5found in the region which underwent deletion were investigated in FAP patients. In the case of four unrelated FAP patients, four mutations were observed in gene DP2.5 (currently – gene APC) which led to the development of a Stop codone from which one was transferred to progeny (1). In the following year 79 FAP patients were examined and mutations in the APC gene were observed in 67% of them. In 92% of cases, they were mutations leading to the truncation of the APC gene protein product (7). In many countries, investigations were undertaken to study the occurrence of mutations in the APC gene and a database of inherited and somatic mutations was established in which data of about 826 inherited and 650 somatic mutations were collected (8). Beginning from 1993, the function of the APC protein was investigated when it was observed to bind with β-catenin which indicated participation in cell adhesion (8).
In Poland, DNA Bank of FAP patients was established in 1997 at the Institute of Human Genetics (IHG) of the Polish Academy of Sciences (PAS) in Poznań. Over 1000 DNA samples were collected in the IHG PAS in Poznań within the framework of investigations concerning genetic succesibility for the development of neoplastic diseases associated with the presence of polyps in the gastrointestinal tract and it is at the IHG that research on familial intestinal polyposis was conducted the longest. At the present time, the Bank stores DNA from 501 FAP patients derived from 400 families. Mild course of the disease was recorded in 11.4% cases of the examined families. Moreover, material from risk groups in which no symptoms of the disease were observed at the moment of qualification for investigations was also gathered. The performed molecular studies made it possible to diagnose mutations in the APC gene in 194 families. The risk group comprised the total of 199 persons in the diagnosed families and for these persons molecular diagnostics was carried out. The examinations performed in the group revealed that no symptoms of the disease were observed in 147 persons. The APC gene mutation was excluded in 97 persons, whereas in 53 persons presymptomatic occurrence of the disease was confirmed.
APC gene structure and APC protein function
The APC ( adenomatous polyposis coli) gene is located on chromosome 5 in the q21 region and contains 21 exons (9). A characteristic attribute of the APC gene is the occurrence of a large exon 15 which encompasses over 70% of the coding sequence. The expression of the gene is observed in all tissues and the transcription product is 8538 nucleotide long mRNA chain (1, 10). First exons can form tissue-specific alternative transcripts (11), e.g. an APC gene product can be found in the brain for which the Start codone is located in the BS ( brain specific) exon. Elimination of codone 1, which encodes the super helix domain, and which is known to serve for homo- or heterodimerisation, affects protein product functionality (9). Alternative assembly of gene initial exons may be associated with the occurrence of the attenuated adenomatous polyposis coli (AAPC) (12). A similar effect was recorded in one of two families with mutations at the 3' end of exon 9 where, in one case, a modifying impact of alternative assembly was observed leading to a mild FAP form. That is why it is believed that the kind, location of mutation as well as the effect of alternative assembly exerts influence on the disease course.
Another very interesting phenomenon is intergenic assembly in the result of which exon 14 as well as exon 15, which comprises nearly 70% of the gene, are removed from the APC gene and the remaining fragment binds with the 3' end of the SRPI gene. Excising exon 14 or 15 results in the development of two product isoforms differing from each other by the ability to bind microtubules and â-catenin as well as by the 3' region which does not undergo translation which can affect mRNA stability and product function (13). An alternative gene assembly, in this case, is associated with the regulation of the APC protein activity and appears to suggest that it fulfils a number of different functions in the cell, especially, that in alternative assembly, over 75% of exons take part.
The full-length APC protein contains 2843 amino acids and can be found in cytoplasm as well as in cell nucleus (1, 10). Several proteins have been recognised which can interact with the APC protein. These proteins include: DLG protein, microtubule protein, GSKβ-3, β-catenin, γ-catenin, p34, Tid56, Auxin protein. Interactions with many proteins indicate that the APC protein participates in the regulation of many cell processes including: division, migration, adhesion and cell fate determination (1). A number of functional domains were identified in the APC protein. The base domain encompasses amino acids 2200-2400 (1). The N-end of protein, between amino acids 1 – 171, is involved in oligomerisation. There are two β-catenin binding sites in the APC protein – in the fragment comprising three 15-nucleotide repeats between amino acids 1020 and 1169 and in the region of 20-amino acid repeats between amino acids 1324 and 2075. Binding with microtubules, which occurs during the increased gene expression, takes place with the assistance of the fragment encompassing amino acids 2130-2483. Amino acids 2450-2843 bind with the EB1 protein, while amino acids 2771-2843 bind with the DLG protein (1). Attempts to identify the region associated with the apoptosis process failed, although it was observed that the expression of the appropriate APC protein in the intestinal neoplastic line cells leads to the occurrence of the phenomenon. A product of the APC gene of 300 kDa mass participates in the contact inhibition of cell growth in mucous cells of the large intestine.
Moreover, APC protein also interacts with γ-catenin and β-catenin. Both proteins bind with an adhesive protein – E-cadherin. Fearon (1997) proposed a model in which the APC protein participates in signal transduction and via β-catenin degradation affects the activity of the Tcf4 ( T-cell transcription factor 4) transcription factor (fig. 1) (14). The protein that regulates the formation of the APC protein and β-catenin complex is protein kinase ZW3/GSK3β. Phosphorilation of the APC protein activates β-catenin binding. The activity of GSK3β kinase is regulated by dsh protein which interacts with the products of gene WNT1. APC protein bound with kinase ZW3//GSK3β is capable of inhibiting the transcription induced by β-catenin. In the case of loss of the APC gene product function, transcription factor Tcf4 ( TCF7L2) becomes activated. The cell is stimulated to proliferation following activation of the transcription of gene c-MYC by Tcf4 (15). The product of the c-MYC gene occurs in the cell nucleus and is capable of binding with DNA, activates growth gene – ornithine decarboxylase ( ODC1) as well as CDC25A gene and, in addition, it inhibits gene GAS1. It was further demonstrated that the activated β-catenin-Tcf4 complex induces Tcf1 expression (16). In the large intestine mucous cells the APC gene serves as a negative regulator of the cell cycle via the regulation of the β-catenin level, a proliferation signal derived from the transmembrane protein E-cadherin. When this gene loses its functionality, the balance between cell divisions and apoptosis in the tissue becomes disturbed.
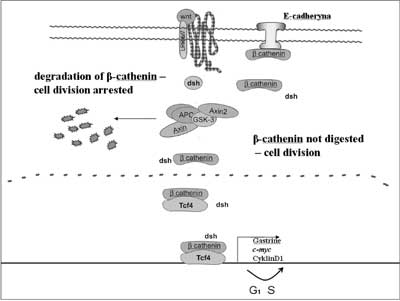
Fig. 1. Role of the APC protein in cell cycle regulation.
Mutation of the APC gene
Development of molecular studies was triggered off in 1991 by the discovery of a relationship between APC gene mutation and FAP. During the period of the past 17 years, the development of various techniques followed by increased numbers of publications was observed in a growing number of countries and ethnic groups connected with the spectrum of mutations. Investigations carried out for several years using such techniques as: the single-stranded conformational polymorphism analysis of DNA fragments (SSCP), heteroduplex (HD) analysis or denaturing gradient gel electrophoresis (DGGE) method as well as DNA sequencing allowed the establishment of computer databases. At the present time, the best APC gene mutation database can be found at the Institute of Medical Genetics in Cardiff http://www.hgmd.cf.ac.uk/ac/index.php as it provides the most up-to-date base of mutations for the majority of described genes (17). At present, information about 858 types of the APC gene mutations have been gathered in the above database. Small deletions constitute the highest proportion of the APC gene mutations. The total of 356 cases have been described and, in majority, they lead to a change in the reading frame and shortening of the gene protein product. Much rarer are large deletions (54), mutations at splice sites (49), small deletions combined with insertions (17), large insertions (7), complex rearrangements (6) and 3 mutations were observed in regulatory sequences. 235 mutations were represented as substitutions leading to changes in amino acids or to the development of the Stop codone at the mutation site. The total of 131 small insertions has been described so far. In the case of substitutions, which constitute 30% of all mutations, the Stop codone arises at the mutation site, whereas in the case of deletions or insertions, which constitute 68% of mutations, the Stop codone arises in its vicinity following the change of the reading frame. There is a region in the gene characterised by increased frequency of mutation occurrence called MCR ( mutation cluster region) which encompasses codones 1055-1309 and 23% of all germinal mutations take place in this region. In addition, among germinal mutations, increased frequency of the following three mutations were observed: deletions of 5 bp in codone 1309 (10%), deletions of 5 bp in codone 1061 and deletions of 4 bp in codone 1064 (18). Majority of mutations can be found in the 5' region of exon 15 of the APC gene. A characteristic FAP attribute is loss of heterozygosity (LOH) in the APC gene developing as a result of somatic mutation in its second allele. The frequency distribution of these mutations differs from germinal mutations. Up to 60% of somatic mutations are situated in the fragment which encompasses 8% of the gene between codones 1286 and 1513. Somatic mutations occur in two hot spots in codone 1309 and codone 1450. Mutations in the APC gene also occur in cases of large intestinal tumours unrelated with polyposis. In cases of these neoplasms, single tumours occur as a result of a mutation in one of the APC gene alleles followed by a loss of heterozygosity in the result of somatic mutations. In the case of the Lynch syndrome (HNPCC), the process is accelerated by inherited mutations in DNA repair genes (19). According to the latest investigations, it is not necessary for LOC to occur in APC in order for the initiation of the large intestine neoplasm to take place. In half of sporadic neoplasms in which no changes in APC took place, tumour development is associated with heterozygotic mutations in the β-catenin gene ( CTNNB1). Mutations occur at the β-catenin phosphorilation site by GSK3β kinase. They cause switching off of the APC gene regulatory function leading to β-catenin accumulation and, hence, expression of the MYC gene and development of the large intestine neoplasm (20). β-catenin occurs downstream of the APC gene along the pathway contact growth inhibition and, in the case of mutations in this gene, the neoplastic process is independent of the condition of the APC gene.
An attempt was made to determine a correlation between the position of translation-terminating mutations and the clinical course of the disease as well as the occurrence of parenteral symptoms. It was found that the occurrence of the Stop codone before the 157 codone decreased the number of polyps and resulted in a mild course of the disease. A classical course of the polyposis with numerous polyps takes place when the Stop signal occurs between codones 169 and 1600. Mutations found between codones 1403 and 1587 lead to the intensification of parenteral symptoms. Occurrence of desmoid tumours is associated with mutations in the region between codones 1445 and 1578. Mutations at the 3' end of the APC gene result in differing picture of the course of the disease both with regard to the polyp numbers and the occurrence of parenteral symptoms.
Progression of morphological and genetic changes in familial adenomatous polyposis
The inheritance of the mutated allele of the APC gene does not result in a clinical picture of the disease. The appearance of clinical symptoms is associated with the sequence of further events in the cell. Both in the case of FAP and HNPCC, cells are characterised by a very high risk of occurrence of the heterogeneity loss in the APC gene locus. In the course of tumour development, changes take place within suppressor, onco- and DNA repair genes. Deletions are observed to occur in regions of the occurrence of proliferation inhibition genes located on 5q ( APC, MCC), 17p ( TP53) and 18q ( DCC) chromosomes, while point mutations are found within the K-ras prooncogene. In the case of FAP, the risk of heterozygosity loss is very high because one of the alleles is inherited in a mutated form. The risk of initiation is also high as a result of mutation inheritance. Despite different causes, the first phase of neoplastic disease initiation is the loss of heterozygosity in the APC locus resulting in increased cell proliferation following the transcription activation of the c-MYC prooncogene. Frequent cell divisions lead to the selection of a clone with an impaired MCC gene which results in dysplasia and, at a later stage, development of the first degree adenoma. Increased rate of cell division causes further accumulation of genetic errors and this results in the activation of the K-ras prooncogene and DCC ( deleted in colorectal cancer) deletion. The successive stage of tumour development is productivity loss of the TP53gene leading to the formation of adenocarcinoma, whereas further error accumulation results in the formation of an invasive tumour. The difference in molecular backgrounds of FAP and HNPCC causes that single tumours in HNPCC develop faster into invasive forms than in the case of FAP.
Structure and location of the MUTYH gene
The MUTYH gene ( MutY, e.coli, Homolog) is a homologue of a bacterial mutY gene involved in the system of oxidative DNA damages. Alternatively, the MUTYH gene is frequently referred to as hMYH or MYH but the above names are considered as inappropriate. In man, the MUTYH gene was described in 1996. It encompasses 7100 bp on chromosome 1 in the region of p34.3-p32.1 and consists of 16 exons. None of the 15 introns of the MYH gene exceed 200 bp. The gene encodes a 535-amino acid protein which is in 41% identical with the mutY protein from E. coli (21). Most frequently, two substitutions Y165C and G382D occur which are observed in 80% of patients with mutations of both gene alleles (22-24).
Phenotype mutation in the APC gene
Familial adenomatous polyposis (FAP) (MIM 175100)
Familial polyposis of the large intestine (FAP) is a predisposition inherited in an autosomal dominant manner for the occurrence of numerous polyps in the large intestine. Symptoms in the form of numerous (hundreds or thousands) polyps in the mucous of the large intestine appear towards the end of the second decade of life. Average age of polyp incidence is 15 years of life (25). Polyps are characterised by high potentials for the development into malignant carcinoma. Risks of neoplasm occurrence in FAP patients reach practically 100%. Malignant tumours may appear from late childhood to the age of 70. The earliest appearance of polyposis symptoms were observed at the age of 5 (26). First symptoms of the disease include: diarrhoea and blood in the stool followed by loss of body weight and general weakness. The disease incidence amounts to 1 in 10 000 cases of newborns (25). Classical form of intestinal polyposis is characterised by the occurrence of over 100 polyps which appear most frequently in the second decade of life as adenomatous polyps and can co-occur with lymphoid polyps (27). The most frequent polyps are tubular ones with diameters reaching even several centimetres. Tubular-villous and villous polyps are also quite frequent (28). Moreover, intestinal polyps can also occur as flat tubular polyps (29). One of the classical forms of polyposis is a phenotype with a severe course (severe FAP) in which the number of polyps exceed one thousand (30). Severe FAP is characterised by earlier age of occurrence of the neoplasm in the intestine, on average 34 years of age, and more frequent occurrence of parenteral symptoms.
Parenteral symptoms in patients suffering from FAP can vary considerably and can include:
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Groden J, Thliveris A, Samowitz W et al.: Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991; 66 (3): p. 589-600.
2. Bisgaard ML, Fenger K, Bülow S et al.: Familial adenomatous polyposis (FAP): frequency, penetrance, and mutation rate. Hum Mutat 1994; 3 (2): p. 121-5.
3. Lynch HT, Smyrk TC: Identyfying Hereditary Nonpolyposis Colorectal Cancer. N Engl J Med 1998; 338: p. 1537-1538.
4. Distante S, Nasioulas S, Somers GR et al.: Familial adenomatous polyposis in a 5 year old child: a clinical, pathological, and molecular genetic study. J Med Genet 1996; 33 (2): p. 157-60.
5. Knudson AJR: Mutation and cancer: statistical study of retinoblastoma. Proc Nat Acad Sci 1971; 68: p. 820-823.
6. Herrera L, Kakati S, Gibas L et al.: Gardner syndrome in a man with an interstitial deletion of 5q. Am J Med Genet 1986; 25 (3): p. 473-6.
7. Miyoshi Y, Nagase H, Ando H, et al.: Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet 1992; 1 (4): p. 229-33.
8. Rubinfeld B, Souza B, Albert I et al.: Association of the APC gene product with beta-catenin. Science 1993; 262 (5140): p. 1731-4.
9. Santoro IM, Groden J: Alternative splicing of the APC gene and its association with terminal differentiation. Cancer Res 1997; 57 (3): p. 488-94.
10. Kinzler KW, Nilbert MC, Su LK et al.: Identification of FAP locus genes from chromosome 5q21. Science 1991; 253 (5020): p. 661-5.
11. Lambertz S, Ballhausen WG: Identification of an alternative 5' untranslated region of the adenomatous polyposis coli gene. Hum Genet 1993; 90 (6): p. 650-2.
12. Samowitz WS, Thliveris A, Spirio LN et al.: Alternatively spliced adenomatous polyposis coli (APC) gene transcripts that delete exons mutated in attenuated APC. Cancer Res 1995; 55 (17): p. 3732-4.
13. Sulekova Z, Reina-Sanchez J, Ballhausen WG: Multiple APC messenger RNA isoforms encoding exon 15 short open reading frames are expressed in the context of a novel exon 10A-derived sequence. Int J Cancer 1995; 63 (3): p. 435-41.
14. Fearon ER: Human cancer syndromes: clues to the origin and nature of cancer. Science 1997; 278 (5340): p. 1043-50.
15. He TC, Sparks AB, Rago C et al.: Identification of c-MYC as a target of the APC pathway. Science 1998; 281 (5382): p. 1509-12.
16. Roose J, Huls G, van Beest M et al.: Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science 1999; 285 (5435): p. 1923-6.
17. Stenson PD, Ball EV, Mort M et al.: Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 2003; 21 (6): p. 577-81.
18. Mandl M, Paffenholz R, Friedl W et al.: Frequency of common and novel inactivating APC mutations in 202 families with familial adenomatous polyposis. Hum Mol Genet 1994; 3 (1): p. 181-4.
19. Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990; 61 (5): p. 759-67.
20. Morin PJ: beta-catenin signaling and cancer. Bioessays 1999; 21 (12): p. 1021-30.
21. Slupska MM, Baikalov C, Luther WM et al.: Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J Bacteriol 1996; 178 (13): p. 3885-92.
22. Sieber OM, Lipton L, Crabtree M et al.: Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med 2003; 348 (9): p. 791-9.
23. Al-Tassan N, Eisen T, Maynard J et al. Inherited variants in MYH are unlikely to contribute to the risk of lung carcinoma. Hum Genet 2003; 114 (2): p. 207-10.
24. Jones S, Emmerson P, Maynard J et al.: Biallelic germline mutations in MYH predispose to multiple colorectal adenoma and somatic G:C -> T:A mutations. Hum Mol Genet 2002; 11 (23): p. 2961-7.
25. Cruz-Correa M, Giardiello FM: Familial adenomatous polyposis. Gastrointest Endosc 2003; 58 (6): p. 885-94.
26. Krush AJ, Traboulsi EI, Offerhaus JA et al.: Hepatoblastoma, pigmented ocular fundus lesions and jaw lesions in Gardner syndrome. Am J Med Genet 1988; 29 (2): p. 323-32.
27. Giardiello FM, Yang VW, Hylind LM et al.: Primary chemoprevention of familial adenomatous polyposis with sulindac. N Engl J Med 2002; 346 (14): p. 1054-9.
28. Giardiello FM, Hamilton SR, Krush AJ et al.: Treatment of colonic and rectal adenomas with sulindac in familial adenomatous polyposis. N Engl J Med 1993; 328 (18): p. 1313-6.
29. Cohen M, Thomson M, Taylor C et al.: Colonic and duodenal flat adenomas in children with classical familial adenomatous polyposis. Int J Surg Pathol 2006; 14 (2): p. 133-40.
30. Cetta F, Dhamo A: Inherited multitumoral syndromes including colorectal carcinoma. Surg Oncol 2007; 16 Suppl 1: p. S17-23.
31. Caspari R, Friedl W, Böker T et al.: Predictive diagnosis in familial adenomatous polyposis: evaluation of molecular genetic and ophthalmologic methods. Z Gastroenterol 1993; 31 (11): p. 646-52.
32. Wallis YL, Macdonald F, Hultén M et al.: Genotype-phenotype correlation between position of constitutional APC gene mutation and CHRPE expression in familial adenomatous polyposis. Hum Genet 1994; 94 (5): p. 543-8.
33. Jagelman DG: Familial Polyposis coli: the clinical spectrum. Prog Clin Biol Res 1988; 279: p. 169-75.
34. Domizio P, Talbot IC, Spigelman AD et al.: Upper gastrointestinal pathology in familial adenomatous polyposis: results from a prospective study of 102 patients. J Clin Pathol 1990; 43 (9): p. 738-43.
35. Kashiwagi H, Kanazawa K, Koizumi M et al.: Development of duodenal cancer in a patient with familial adenomatous polyposis. J Gastroenterol 2000; 35 (11): p. 856-60.
36. Zwick A, Munir M, Ryan CK et al.: Gastric adenocarcinoma and dysplasia in fundic gland polyps of a patient with attenuated adenomatous polyposis coli. Gastroenterology 1997; 113 (2): p. 659-63.
37. Sarre RG, Frost AG, Jagelman DG et al.: Gastric and duodenal polyps in familial adenomatous polyposis: a prospective study of the nature and prevalence of upper gastrointestinal polyps. Gut 1987; 28 (3): p. 306-14.
38. Heiskanen I, Kellokumpu I, Jarvinen H: Management of duodenal adenomas in 98 patients with familial adenomatous polyposis. Endoscopy 1999; 31 (6): p. 412-6.
39. Jones IT, Jagelman DG, Fazio VW et al.: Desmoid tumors in familial polyposis coli. Ann Surg 1986; 204 (1): p. 94-7.
40. Klemmer S, Pascoe L, DeCosse J: Occurrence of desmoids in patients with familial adenomatous polyposis of the colon. Am J Med Genet 1987; 28 (2): p. 385-92.
41. Camiel MR, Mulé JE, Alexander LL et al.: Thyroid carcinoma with Gardner's syndrome. N Engl J Med 1968; 279 (6): p. 326.
42. Bell B, Mazzaferri EL: Familial adenomatous polyposis (Gardner's syndrome) and thyroid carcinoma. A case report and review of the literature. Dig Dis Sci 1993; 38 (1): p. 185-90.
43. Brozek I, Plawski A, Podralska M et al.: Thyroid cancer in two siblings with FAP syndrome and APC mutation. Int J Colorectal Dis 2008 23 (3): p. 331-2.
44. Itoh H, Ohsato K: Turcot syndrome and its characteristic colonic manifestations. Dis Colon Rectum 1985; 28 (6): p. 399-402.
45. Itoh H, Ohsato K, Yao T et al.: Turcot's syndrome and its mode of inheritance. Gut 1979; 20 (5): p. 414-9.
46. Paraf F, Jothy S, Van Meir EG: Brain tumor-polyposis syndrome: two genetic diseases? J Clin Oncol 1997; 15 (7): p. 2744-58.
47. Van Meir EG: „Turcot's syndrome”: phenotype of brain tumors, survival and mode of inheritance. Int J Cancer 1998; 75 (1): p. 162-4.
48. Giardiello FM, Petersen GM, Brensinger JD et al.: Hepatoblastoma and APC gene mutation in familial adenomatous polyposis. Gut 1996; 39 (96): p. 867-9.
49. Gruner BA, DeNapoli TS, Andrews W et al.: Hepatocellular carcinoma in children associated with Gardner syndrome or familial adenomatous polyposis. J Pediatr Hematol Oncol 1998; 20 (3): p. 274-8.
50. Bishop DT, Thomas HJ: The genetics of colorectal cancer. Cancer Surv 1990; 9 (4): p. 585-604.
51. Soravia C, Berk T, Madlensky L et al.: Genotype-phenotype correlations in attenuated adenomatous polyposis coli. Am J Hum Genet 1998; 62 (6): p. 1290-301.
52. Nieuwenhuis MH, Vasen HF: Correlations between mutation site in APC and phenotype of familial adenomatous polyposis (FAP): a review of the literature. Crit Rev Oncol Hematol 2007; 61 (2): p. 153-61.
53. Sampson JR, Dolwani S, Jones S et al.: Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet 2003; 362 (9377): p. 39-41.
54. Farrington SM, Tenesa A, Barnetson R et al.: Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am J Hum Genet 2005; 77 (1): p. 112-9.
55. Barnetson RA, Devlin L, Miller J et al.: Germline mutation prevalence in the base excision repair gene, MYH, in patients with endometrial cancer. Clin Genet 2007; 72 (6): p. 551-5.
56. Nielsen M, Hes FJ, Nagengast FM et al.: Germline mutations in APC and MUTYH are responsible for the majority of families with attenuated familial adenomatous polyposis. Clin Genet 2007; 71 (5): p. 427-33.
57. Mastronardi L, Ferrante L, Lunardi P et al.: Association between neuroepithelial tumor and multiple intestinal polyposis (Turcot's syndrome): report of a case and critical analysis of the literature. Neurosurgery 1991; 28 (3): p. 449-52.
58. Plawski A, Slomski R: Geny Supresorowe Nowotworów. Postępy Biologii Komórki 1998; 25 (sup. 10): p. 251-264.
59. Plawski A, Nowakowska D, Podralska M: The AAPC case, with an early onset of colorectal cancer. Int J Colorectal Dis 2007; 22 (4): p. 449-51.
60. Skrzypczak M, Podralska M, Heinritz W et al.: The MYH gene status in Polish FAP patients without the APC gene mutations. Hereditary Cancer in Clinical Practice 2006; 4: p. 43-47.
61. Kubinska I et al.: Using of molecular sreening in children with familial polyposis background – own experience. Polish Journal of Environmental Studies 2006; 15 (5b): p. 98-101.
62. Plawski A, Lubiński J, Banasiewicz T et al.: Novel germline mutations in the adenomatous polyposis coli gene in Polish families with familial adenomatous polyposis. J Med Genet 2004; 41 (1): p. e11.
63. Plawski A, Slomski R: The APC gene mutations causing FAP in Polish patients. Journal of Applied Genetics 2008; 49 (4): p. 407-414.
64. Nagel R, le Sage C, Diosdado B et al.: Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res 2008; 68 (14): p. 5795-802.
65. Oshima M, Dinchuk JE, Kargman SL et al.: Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996; 87 (5): p. 803-9.
66. Chiu CH, McEntee MF, Whelan J: Sulindac causes rapid regression of preexisting tumors in Min/+ mice independent of prostaglandin biosynthesis. Cancer Res 1997; 57 (19): p. 4267-73.
