© Borgis - Postępy Nauk Medycznych 7/2010, s. 524-534
Dagmara Dymerska, Pablo Serrano-Fernández, Joanna Trubicka, Bartłomiej Masojć, *Grzegorz Kurzawski
DNA and RNA analyses in detection of genetic predisposition to cancer
Analizy molekularne DNA i RNA w wykrywaniu dziedzicznych predyspozycji do nowotworów
International Hereditary Cancer Centre, Department of Genetics and Pathology, Pomeranian Medical University, Szczecin, Poland
Head of Department of Genetics and Pathology: prof. zw. dr hab. med. Jan Lubiński
Streszczenie
W ostatnich latach obserwuje się dynamiczny rozwój metod molekularnych służących do analiz DNA i RNA. Niegdyś powszechnie stosowane techniki takie jak SSCP, HET, CMC, DGGE, RFLP czy ASA są stopniowo wypierane przez nowe, umożliwiające tańsze i szybsze diagnozowanie pacjentów. Aktualnie prym wiodą metody oparte na PCR w czasie rzeczywistym oraz metody umożliwiające multipleksację kilku reakcji. Nowe metody skriningowe pozwalają na badanie dużych grup pacjentów stosunkowo niskim kosztem. Pojawienie się wielofunkcyjnych robotów używanych w izolacji, normalizacji DNA, przygotowaniach reakcji PCR itp., zrewolucjonizowało pracę w laboratorium, czyniąc ją łatwą i przyjemną przy równoczesnym ograniczeniu ryzyka zanieczyszczenia i kontaminacji prób.
W niniejszym opracowaniu przedstawiono nowe, dostępne w standardowych laboratoriach metody stosowane w wykrywaniu mutacji konstytucyjnych u osób z genetyczną predyspozycją do nowotworów.
Summary
During the past decade many new molecular methods for DNA and RNA analysis have emerged. The most popular in the past like SSCP, HET, CMC, DGGE, RFLP or ASA have been replaced by methods which allowed more cost effective and less time consuming testing. Real-time techniques and particularly those ones with capability of high multiplexing have become commonly used in laboratory practice. Novel screening methods enable examining large series of patients in short time. Use of handling liquid robots, their application in DNA or RNA isolation, normalisation of samples concentration, PCR preparation, etc. have reduced risk of contamination and have made laboratory work much easier and faster.
The aim of this study was the introduction of a few modern techniques, most commonly used in detection of genetic predisposition to cancer.

Several genes have been identified which, if mutated, are associated with increased predisposition to tumours (1).
Carriers of mutations in these genes show a risk of cancer from a few percent to as high as 90%. Selected genes associated with high inherited predisposition to tumours and those most frequently examined in clinical practice are summarised in table 1 (2-6).
Table 1. Genes associated with predisposition to cancer family syndromes. The table contains genes studied the mostly frequently in our centre.
| GENE LOCALISATION | PREDISPOSITION TO MALIGNANCIES | PENETRANCE* |
Rb1 [2]
13q14 | retinoblastoma | about 90% |
BRCA1 [3]
17q21 | breast cancer
ovarian cancer | about 80% |
BRCA2 [3]
13q13 | prostate cancer
colon cancer | |
VHL [4]
3p25 | haemangioblastoma of the cerebellum and retina kidney cancer pheochromocytoma | about 80% |
MSH2 [5]
2p21 | colon cancer endometrial cancer | about 90%
for male
about 70%
for female [6] |
MLH1 [5]
3p22 | cancer of the stomach, cancer of the biliary |
MSH6 [5]
2p16 | tract small bowel cancer ovarian cancer |
*probability of malignancy during lifetime among mutation carriers
Several molecular methods have been devised aimed at detecting mutations. They can be subdivided into methods detecting:
? new mutations,
? known mutations.
DETECTION OF NEW MUTATIONS
Diagnostics of such mutations in cases appropriately preselected using pedigree and clinical data is justified in clinical practice, even though such techniques are still complex, time-consuming and expensive.
DNA analyses
The main kinds of analyses:
? DNA isolation,
? amplification of gene fragments, usually of coding sequences,
? preliminary detection of changes within amplification products using screening techniques,
? sequencing and pyrosequencing,
? southern method and multiplex ligation-dependent probe amplification,
? high resolution melting analysis.
DNA isolation
Material for DNA isolation is usually taken from blood leukocytes or less frequently from other tissues. Analyses allow detection of mutations which are constitutional, i.e. present in all cells of the patients. It is especially effective if the biological material for analysis is collected immediately before isolation. However, good results can be achieved even after a few days of storage of blood at room temperature or even storage for a few years at temperature below zero. If fresh tissue is not available, DNA isolation can be performed from tissues fixed in formalin and embedded in paraffin blocks, although the achievement of unequivocal results using such material is more difficult and sometimes even impossible. DNA isolation requires elimination of proteins from cellular lysate. In the phenol-chloroform method this is achieved by digestion with proteinase K and extraction in a mixture of phenol and chloroform. Finally, nuclear acids are extracted using ethyl- or isopropyl- alcohols. At present, the above technique is used only exceptionally although it produces clean and non-degraded DNA (in practice it is used only for DNA isolation from paraffin blocks). Instead, other techniques are used which are less laborious and easier for automation. They are based on selected labelling of DNA with template (e.g. with dyne beads) and then washing in order to separate the DNA.
Amplification of gene fragments
DNA fragments are amplified using polymerase chain reaction. The reaction mixture includes: DNA template (usually genomic DNA), DNA polymerase, a pair of specific primers, deoxyribonucleotide triphosphates and the reaction buffer. This mixture is exposed to cyclic changes of temperature. Each cycle includes: denaturation, starters annealing and synthesis. After 22 cycles, assuming 100% effectiveness, the copy number of the amplified fragment is increased one million-fold.
Preliminary detection of changes within amplification products using screening techniques
DNA-SSCP (single strand conformational polymorphism) was in the past the most popular technique of initial detection of changes in amplification products (7).
Other techniques of this kind include HET (heteroduplex analyses) (8), CMC (chemical mismatch cleavage) (9), DHPLC (denaturing high-performance liquid chromatography) (10) and DGGE (denaturing gradient gel electrophoresis) (11).
DHPLC (denaturing high-performance liquid chromatography)
At present, the best and the most frequently applied technique of initial detection of changes is DHPLC (10, 12-15). This is a kind of HET based on high resolution of modern chromatographic columns. Analysed DNA fragments are separated in a gradient of denaturing agent. (The key to DHPLC is the solid phase, which has differential affinity for single and double-stranded DNA). Under sub-denaturing conditions, heteroduplexes show lower affinity than homoduplexes to the solid phase of the column and it is easier to elute them. Separation is monitored by UV absorption measured at 260 nm. The elution profile (fig. 1) is characteristic and replicable for a given change and allows differentiation between new changes and already known mutations or polymorphisms.
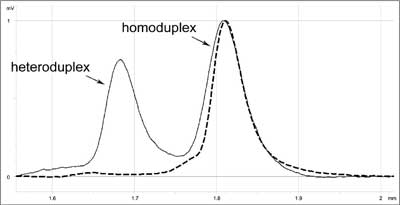
Fig. 1. DHPLC elution profile characteristic for c.1786_1788delAAT mutation in MSH2 (mutS homolog 2) gene (solid line) compared to 'wild' type (dashed line).
Based on literature data (16) and our own experience (17) we can say that DHPLC combines the advantages of several methods. Its sensitivity is close to 100% (10, 14, 15). At the same time the cost is relatively low (cost of reagents per sample is 5-10 Euro). The method is quick, and if an autosampler is used, it allows 200 samples to be analysed per day.
Sequencing
Sequencing is the most sensitive technique for detection of changes in genomic material, allowing at the same time their full characterisation.
In the nineties, significant progress in sequencing technologies has been achieved by application of automated machines, for which the identification of particular nucleotides is based on fluorescence induced by laser. Each nucleotide (A, C, G, T) can be labelled with a different fluorescent dye. The most convenient technique is the cyclic sequencing method (18).
During the analysis the sequences of PCR products for both DNA strands are assessed. The real change is detected in both DNA strands. The sequencing procedure comprises several stages:
? preparative PCR – based on the amplification of a chosen fragment of the gene using pairs of specific starters,
? asymmetric PCR – separate amplification with each of the starters using fluorescent dye-labelled dideoxynucleotides,
? electrophoresis in denaturing polyacrylamide gel with simultaneous detection and registration of products,
? analysis of the results using computer programs.
During asymmetric PCR all possible oligonucleotides of different length, complementary to the template and containing fluorochromes at the 3'-end are created. They are separated during electrophoresis and the particular order of coloured nucleotides can be read as the sequence complementary to the template. The detected DNA sequence is compared with the wild sequence (fig. 2) available in databases such as GenBank and EMBL, and the type of change can be precisely described.
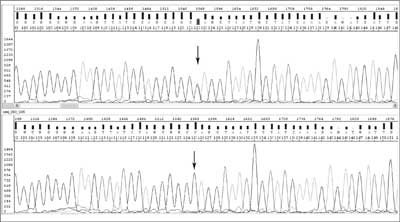
Fig. 2. Chromatograms for DNA sequencing: studied sequence with c.83C>T mutation in MLH1 (mutL homolog 1) gene (upper) and 'wild' sequence (below).
Currently, the leading companies are offering modern automated DNA sequencing instruments (DNA sequencers) allowing simultaneous sequencing of 96 samples based on capillary electrophoresis of products using the cyclic method using fluorescent dye-labelled dideoxynucleotides. Recent progress in this discipline has based not only on increasing the number of simultaneously analysed samples but also on improved "chemistry” such as gel compositions, which enable accurate sequencing of almost one thousand bases of one fragment.
New sequencers ("GS FLX machines”) rely on real time sequencing by simultaneous synthesis of many short DNA fragments (around 400 hundred base pairs in length). They apply pyrosequencing with detection of luminescence occurring during ATP degradation. These machines allow analyses of 400-600 million base pairs per instrument run. Pyrosequencing (19, 20) uses an one strand DNA fragment as the template on which synthesis of a complementary strand is performed through addition of 4 different deoxynucleotides triphosphate (dNTPs). Addition of each base is associated with liberation of pyrophosphate which is transformed into ATP using sulfurylase and adenosine-5'-phosphosulfate. ATP is used by luciferase for the transformation of luciferin into oxyluciferin. During this reaction light is generated with an intensity corresponding to the amount of pyrophosphate produced early. This light is registered by CCD and transformed into peaks on a pyrogram (fig. 3). The same reaction scheme is valuable for different dNTPs. If the added nucleotide is not complementary to the template it is not included in the newly synthesised strand and pyrophosphate is not created. The presence of a light signal is the requisite for adding a new nucleotide to a given sequence.

Fig. 3. Pyrograms: patient with c.2932C>T in APC (adematous polyposis coli) gene (upper) and 'wild' type (below).
Based on our own experience the utmost disadvantage of pyrosequencing technique is the difficulty in determining the number of incorporated nucleotides in regions with base pairs repetitions. In homopolymeric regions of more than 5 nucleotides, estimation of complementary joined nucleotides is impossible due to nonlinear light response. However the problem can be solved by using nucleotide reversible terminators (NRTs), analogues modified by attaching a cleavable fluorophore to the base and a chemically reversible moiety to the 3' cap (3'- O -allyl or 3'- O -(2-nitrobenzyl)). During the extension step when the complementary NRT is incorporated, the reaction is temporarily terminated and resumed only when the capping moiety is removed (by deallyation or laser irradiation). In this way, when analysing the results on the pyrogram, each peak correspond to each incorporated nucleotide and homopolymeric regions can be clearly identified (21).
DNA mutation covering the sites of primers annealing or other fragments outside of amplified ones are not detected using DNA tests based on analyses described above. Some of such changes are large rearrangements.
Southern method and MLPA (multiplex ligation-dependent probe amplification)
A technique very popular in the past for detection of large rearrangements was Southern blotting, described for the first time by E. M. Southern in 1975.
At present the method which has almost completely replaced detection of DNA rearrangements by Southern blotting is MLPA (multiplex ligation-dependent probe amplification) (22). This technique is based on the ligation of specific probes and their subsequent amplification and allows an assessment of the exon copy numbers to be made. On this basis, conclusions can be drawn concerning deletions or duplications of gene fragments or of whole genes.
In this technique many probes are used simultaneously in "one tube”. Probes matching the sequences complementary to exon sequences also contain primer sequences and one of each pairs additionally a unique insertion sequence called a stuffer sequence. Hybridising sequences of each pair of probes match neighbouring DNA fragments and only if hybridisation is complete can ligation take place. After probe hybridisation to the template, the DNA fragments are ligated, then denatured. The dissociated ligated probe containing primer sequences is then amplified using PCR. The presence of stuffers of different lengths allows differentiation of products labelling different targets, and the amount of product is proportional to the copy number in the template. Each peak corresponds to the product of amplification of specific ligated pairs of probes (fig. 4). Relative differences in the height or area of the peak indicate quantitative (sometimes qualitative) changes of a target sequence for the probe.
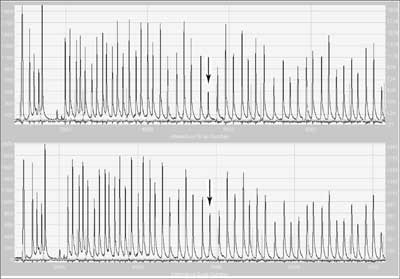
Fig. 4. Result of MLPA electrophoresis of a sample with deletion of exon 9 in MSH2 (mutS homolog 2) gene (upper) and 'wild' type (below).
The advantages of this technique are that only a small amount of DNA is necessary to perform analyses and that efficiently reproducible results may be achieved even from degraded genetic material.
Commercially available probes include those for the most important genes associated with a high risk of tumours, such as: ATM, BRCA1, BRCA2, CHEK1, MLH1, MSH2, MSH6, PMS2, APC, FANCA, FANCD2, PTCH, BMPR1A, SMAD4, TP53, CDH1, MEN1, NF1, NF2, STK11, SMARCB1, RB1, CDKN2A-CDKN2B, WT1.
HRMA (high resolution melting analysis)
This real-time PCR based method can be used for detection of SNPs as well as for large rearrangements. All mutations (small and large) can be screened simultaneously in one test, which allows reducing testing time. The basis of the genotyping is a unique pattern of melting curves.
The first step of the analysis is real-time PCR with fluorescent dye, mostly SYBR Green, LCGreen or Syto 9 (23). That allows monitoring the amplification of the DNA template, since fluorescence intensity is proportional to the amount of double-strand DNA (dsDNA). After overheating, the melting behaviour of the PCR products is monitored by plotting the changes in fluorescence that occur by denaturating double-strand DNA (dsDNA) (fig. 5). The pattern of melting temperature (Tm) differences can allow the discrimination of homo- and heterozygotes. The main problem of the HRMA method is that differences in the melting curve shape can easily identify heterozygotes, but may not distinguish all homozygotes (24). However, high sensitivity and specificity, low costs, small amount of DNA required (<5 ng) and the simplicity of the method are prominent features that make HRMA a great candidate as new screening method for cancer predisposition genes (23, 25).
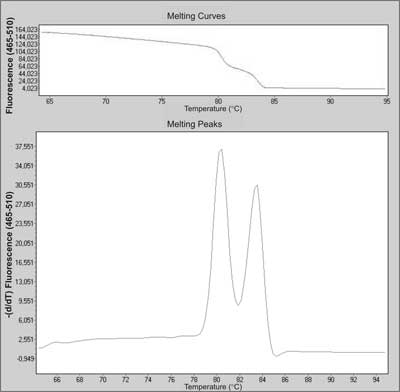
Fig. 5. Melting curve and melting peaks charts of a heterozygous mutation in exon 23 in NF-1 (neurofibromatosis 1) gene during screening by HRMA.
RNA analyses
The advantages of RNA analyses are mainly due to the possibility of detecting a mutation using a lower number of reactions (this is related to the shorter length of RNA in comparison to DNA). So far the main disadvantages of these techniques have included difficulties in achieving reproducible results, lower stability of RNA with mutations and troubles in interpretation associated with occurrence of RNA alternative splicing.
RNA analyses include a few stages:
? RNA isolation,
? amplification of coding parts of the genes,
? detection of changes in amplification products.
RNA isolation
In the majority of laboratories, RNA is isolated from peripheral blood lymphocytes. RNA isolation is performed similarly to DNA isolation. However, due to the widespread presence of thermostable RNAses in tissues, RNA isolation has to be performed more carefully. It is very popular to use the isolation method by Chomczyński (26) from cellular lysates in a solution of guanidine thiocyanate (RNA inhibitors) followed by extraction in a mixture of phenol and chloroform. Slightly acidic pH of phenol leads to extraction of not only proteins but also of DNA, which under such conditions is practically insoluble.
RT-PCR (reverse transcriptase PCR)
RNA can be transcribed into cDNA (complementary DNA) using reverse transcriptase and then amplified using PCR. RNA does not include introns, so only a few pairs of starters usually allow amplification of the whole coding parts of selected genes. Analysing cDNA on regular agarose gels usually allows detection of RNA abnormalities which are caused by deletions or insertions of over a few dozen base pairs or splice mutations.
Detection of alterations in amplification products
Products of the RT-PCR reaction can be analysed using all of the techniques mentioned above and by using the in vitro transcription translation (IVTT) assay, also called the protein truncation test (PTT) (27, 28). RT-PCR is the first stage of PTT. In PTT one primer includes sequences not only initiating transcription into cDNA but also sequences initiating translation – it is protein synthesis " in vitro ” on a cDNA template. After electrophoresis and transmission into membranes, the length of the synthesised protein is assessed, which is altered not only by large deletions or insertions within the RNA but also when single nucleotide mutations lead to stop coding (TGA, TAA or TAG) or splicing mutations. The disadvantage of PTT is its limitation in the detection of missense mutations. It is estimated that even if we use all known tests of direct detection of changes their sensitivity does not exceed 70-80%, mainly due to the lack of diagnosis of alterations in sequences regulating the function.
DETECTION OF KNOWN MUTATIONS
More and more is known about the type and frequency of mutations predisposing to tumours, which are characteristic for different populations, including founder mutations and changes recurrently occurring in families of a given ethnic group. DNA tests aiming detection of all known mutations are highly valuable due to the unusually high economical effectiveness. Such genes as BRCA1, MLH1, MSH2and VHL have been studied intensively in many populations and it is known which mutations should be checked first and in which fragments of genes (29-31).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Lubiński J, Górski B, Kurzawski G et al.: Molecular basis of inherited predispositions for tumors. Acta Biochim Pol 2002; 49: 571-581.
2. Schubert EL, Hansen MF, Strong LC: The retinoblastoma gene and its significance. Ann Med 1994; 26: 177-184.
3. Gronwald J, Menkiszak J, Tołoczko A et al.: Hereditary breast cancer. Pol J Pathol 1998; 49: 59-66.
4. Neumann HP, Zbar B: Renal cysts, renal cancer and von Hippel-Lindau disease. Kidney Int 1997; 51: 16-26.
5. Lynch HT, Smyrk T: Hereditary nonpolyposis colorectal cancer (Lynch syndrome). Cancer 1996; 78: 1149-1167.
6. Dunlop MG, Farrington SM, Carothers AD et al.: Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet 1997; 6: 105-110.
7. Orita M, Iwahana H, Kanazawa H et al.: Detection of polimorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. PNAS 1989; 86: 2766-2770.
8. Nagamine CM, Chan K, Lau YFCA: A PCR artifact: generation of heteroduplexes. Am J Hum Genet 1989; 45: 337-339.
9. Cotton RG, Rodrigues NR, Campbell RD: Reactivity of cytosine and thymine in single-base-pair mismatches with hydroxylamine and osmium tetroxide and its application to the study of mutations. PNAS 1988; 85: 4397-4401.
10. O'Donovan MC, Oefner PJ, Roberts SC et al.: Blind analysis of denaturing high-performance liquid chromatography as a tool for mutation detection. Genomics 1998; 52: 44-49.
11. Myers RM, Maniatis T, Lerman LS: Detection and localization of single base changes by denaturing gradient gel electrophoresis. Methods Enzymol 1987; 155: 501-527.
12. Liu W, Smith DI, Rechtzigel KJ et al: Denaturing high performance liquid chromatography (DHPLC) used in the detection of germline and somatic mutations. Nucleic Acids Res 1998; 26: 1396-1400.
13. Jones AC, Austin J, Hansen N et al.: Optimal temperature selection for mutation detection by denaturing HPLC and comparison to single-stranded conformation polymorphism and heteroduplex analysis. Clin Chem 1999; 45: 1133-1140.
14. Arnold N, Gross E, Schwarz-Boeger U et al.: A highly sensitive, fast and economical technique for mutation analysis in hereditary breast and ovarian cancers. Hum Mutat 1999; 14: 333-339.
15. Gross E, Arnold N, Goette J et al.: A comparison of BRCA1mutations analysis by direct sequencing, SSCP and DHPLC. Hum Genet 1999; 105: 72-78.
16. Xiao W, Oefner PJ: Denaturing high-performance liquid chromatography: A review. Hum Mutat 2001; 17: 439-474.
17. Kurzawski G, Safranow K, Suchy J et al.: Mutation analysis of MLH1and MSH2genes performed by denaturing high-performance liquid chromatography. J Biochem Biophys Methods 2002; 51: 89-100.
18. Rosenthal A, Charnock-Jones DS: New protocols for sequencing with dye terminators. DNA Seq 1992; 3: 61-64.
19. Ronaghi M, Karamohamed S, Pettersson B et al.: Real-time DNA sequencing using detection of pyrophosphate release. Anal Biochem 1996; 242: 84-90.
20. Agah A, Aghajan M, Mashayekhi F et al.: A multi-enzyme model for pyrosequencing. Nucleic Acids Res 2004; 32: e166.
21. Wu J, Zhang S, Meng Q et al.: 3'-O-modified nucleotides as reversible terminators for pyrosequencing. Proc Natl Acad Sci 2007; 104: 16462-16467.
22. Schouten JP, McElgunn CJ, Waaijer R et al.: Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002; 30: e57.
23. Wittwer CT: High-resolution DNA melting analysis: advancements and limitations. Hum Mutat 2009; 30: 857-859.
24. Erali M, Voelkerding KV, Wittwer CT: High resolution melting applications for clinical laboratory medicine. Exp Mol Pathol 2008; 85: 50-58.
25. Rouleau E, Lefol C, Bourdon V et al.: Quantitative PCR high-resolution melting (qPCR-HRM) curve analysis, a new approach to simultaneously screen point mutations and large rearrangements: application to MLH1germline mutations in Lynch syndrome. Hum Mutat 2009; 30: 867-875.
26. Chomczyński P, Sacchi N: Single step method of RNA isolation by acid guanidinum thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162: 156-159.
27. Luce MC, Marra G, Chauhan DP et al.: In vitro transcription/translation assay for the screening of h MLH1 and h MSH2 mutations in familial colon cancer. Gastroenterology 1995; 109: 1368-1374.
28. Plumer SJ, Casey G: Are we closer to genetic testing for common malignances? Nat Med 1996; 2: 156-158.
29. Kurzawski G, Suchy J, Kładny J et al.: Germline MSH2and MLH1mutational spectrum in HNPCC families from Poland and the Baltic States. J Med Genet 2002; 39: E65.
30. Cybulski C, Krzystolik K, Murgia A et al.: Germline mutations in the von Hippel-Lindau ( VHL) gene in patients from Poland: disease presentation in patients with deletions of the entire VHL gene. J Med Genet 2002; 39: E38.
31. Górski B, Byrski T, Huzarski T et al.: Founder mutations in the BRCA1gene in Polish families with breast-ovarian cancer. Am J Hum Genet 2000; 66: 1963-1968.
32. Heied CA, Stevens J, Livak KJ, Williams PM: Real time quantitative PCR. Genome Res 1996; 6: 986-994.
33. Matsubara Y, Fujii K, Rinaldo P, Narisawa K: A fluorogenic allelespecific amplification method for DNA-based screening for inherited metabolic disorders. Acta Paediatr Suppl 1999; 88: 65-68.
34. Haugland RP: The handbook of Fluorescent Probes and Research products. Ninth Edition. Molecular Probes 2002 (http://www.probes.com)
35. Kutyavin IV, Afonina IA, Mills A et al.: 3'-minor groove-binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res 2000; 28: 655-661
36. Wise CA, Paris M, Morar B et al.: A standard protocol for single nucleotide primer extension in the human genome using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom 2003; 17: 1195-1202
37. Gabriel S, Ziaugra L, Tabbaa D: SNP genotyping using the Sequenom MassARRAY iPLEX platform. Curr Protoc Hum Genet 2009, Chapter 2: Unit 2.12.
38. Dymerska D, Serrano-Fernández P, Suchy J et al.: Combined iPLEX and TaqMan Assays to Screen for 45 Common Mutations in Lynch Syndrome and FAP Patients. J Mol Diagn 2010; 12: 82-90.
39. Wang W., Kham SK, Yeo GH et al.: Multiplex minisequencing screen for common Southeast Asian and Indian beta-thalassemia mutations. Clin Chem 2003; 49: 209-218.
40. Révillion F, Verdičre A, Fournier J et al.: Multiplex single-nucleotide primer extension analysis to simultaneously detect eleven BRCA1mutations in breast cancer families. Clin Chem 2004; 50: 203-206.
41. Bujalkova M, Zavodna K, Krivulcik T et al.: Multiplex SNaPshot genotyping for detecting loss of heterozygosity in the mismatch-repair genes MLH1and MSH2in microsatellite-unstable tumors. Clin Chem 2008; 54: 1844-1854.
42. Konstantou JK, Ioannou PC, Christopoulos TK: Dual-allele dipstick assay for genotyping single nucleotide polymorphisms by primer extension reaction. Eur J Hum Genet 2009; 17: 105-111.
43. Litos IK, Ioannou PC, Christopoulos TK et al.: Multianalyte, dipstick-type, nanoparticle-based DNA biosensor for visual genotyping of single-nucleotide polymorphisms. Biosens Bioelectron 2009; 24: 3135-3139.
44. Gialeraki A, Markatos C, Grouzi E et al.: Evaluation of a reverse-hybridization StripAssay for the detection of genetic polymorphisms leading to acenocoumarol sensitivity. Mol Biol Rep 2010; 37: 1693-1697.
