© Borgis - Postępy Nauk Medycznych 7/2010, s. 546-549
*Józef Kładny1, Janina Suchy2, Dagmara Dymerska2, Grzegorz Kurzawski2, Tadeusz Dębniak2, Jan Lubiński2
Lynch Syndrome (HNPCC)
Zespół Lyncha (HNPCC)
1Department of General and Oncological Surgery, Pomeranian Medical University, Szczecin, Poland
Head of Department of General and Oncological Surgery: prof. dr hab. med. Józef Kładny
2International Hereditary Cancer Center, Department of Genetics and Pathology, Pomeranian Medical University, Szczecin, Poland
Head of Department of Genetics and Pathology: prof. zw. dr hab. med. Jan Lubiński
Streszczenie
Praca jest przeglądem doświadczeń własnych oraz danych z piśmiennictwa na temat diagnostyki, profilaktyki i leczenia nowotworów w przebiegu zespołu Lyncha (HNPCC).
Unikalną wartość wnoszą odmienne od standardowych kryteria rozpoznawania „HNPCC susp”. Taką sytuację kliniczną według własnych badań autorów należy rozpoznawać wówczas, gdy:
1. u probanta lub któregokolwiek z jego krewnych I° lub II° stwierdza się raka jelita grubego;
2. u chorego z rakiem jelita grubego, spełniającego kryterium 1) lub wśród jego krewnych I° stwierdza się raka z tzw. spektrum HNPCC – raka jelita grubego, trzonu macicy, jelita cienkiego lub dróg moczowych;
3. co najmniej jeden z raków spełniających kryteria 1) lub 2) zdiagnozowany został poniżej 50. r.ż.
4. wykluczono polipowatość rodzinną.
Summary
Publication is the review based on author's own experience and literature data concerning diagnosis, prevention and treatment of tumors in families with Lynch Syndrome (HNPCC).
The unique value is provided by special, distinct from standard, criteria of diagnosing "HNPCC susp”. According to authors studies such diagnosis can be established when:
1. proband or at least one of his I° or II° relatives is affected by colorectal cancer (CRC);
2. patient with CRC matching criterium 1) or one of his I° relatives is affected by at least one cancer from so called HNPCC spectrum – CRC, cancer of the endometrium or of the small bowel or of the urinary tract;
3. at least one of cancers matching criteria 1) or 2) has been diagnosed under age of 50 years;
4. familial adenomatous polyposis is excluded.

It is estimated that a high genetic predisposition is the cause of 10-20% of all colon cancers (CRC) (1-5).
Among the well known syndromes of inherited predisposition to tumours manifesting with CRC such syndromes showing mendelian pattern of inheritance can be included like: hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome), familial adenomatous polyposis (FAP); Gardner, Zankas, Turcot, Peutz-Jaghers syndromes and juvenile polyposis.
Lynch syndrome (HNPCC)
HNPCC has been described by Lynch (6) in 60's and is the cause of around 5% of all CRC. It has been shown that HNPCC is caused by mutations within several genes such as: MSH2, MLH1, MSH6, EPCAM, PMS2. Mutation within the two first are the most frequent cause of the Lynch syndrome (7-10). Characteristic clinical feature of Lynch syndrome include:
– early age of CRC diagnosis (about 45 yrs),
– more frequent right site tumour localization,
– two and more CRC cases among I° relatives,
– many syn- and meta-chronous CRC tumours
– occurrence of disease in consecutive generations (vertical transmissions)
– increased frequency of occurrence among relatives of cancers of the endometrium, small bowel and urinary tract
According to international group of experts (International Collaborative Group on HNPCC – ICG-HNPCC) Lynch syndrome can be definitively diagnosed, if constitutional mutation within one of genes connected with HNPCC, such as MSH2 or MLH1 is identified or if the following pedigree-clinical criteria are matched (tab. 1) (11, 12).
Table 1. Diagnostic criteria of HNPCC according to ICG-HNPCC (12).
| 1. | at least 3 relatives are affected by histologically verified CRC or cancer of the endometrium, small bowel or urinary tract; at least one of them is I° relative to the other two; FAP is excluded* |
| 2. | at least 2 of above persons are I° relatives from two different generations |
| 3. | at least 1 of above persons with cancer diagnosed at age under 50 yrs |
All other parameters (right site localization, syn- or metachronous tumours) should be treated like non-diagnostic features.*colorectal polyposis, congenital hypertrophy of the retinal pigment epithelium, cysts and osteomas of the mandible/maxilla and desmoids are excluded.
A family matching definitive criteria of HNPCC according to ICG-HNPCC is presented on figure 1.
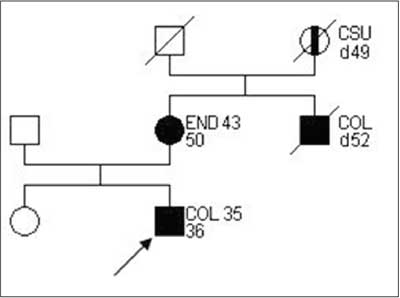
Fig. 1. Family pedigree matching HNPCC criteria, according to ICG-HNPCC.
Due to incomplete penetrance of genes what is typical for dominant mendelian disorders, deaths caused by various diseases, or due to difficulties in achieving full information about all of relatives, the large proportion – perhaps majority – of families actually with HNPCC, can not be diagnosed using Amsterdam criteria summarized in table 1.
Therefore several authors is using another type of criteria, fulfillment of which is not allowing definitive diagnosis of HNPCC, however it is useful in identification of families with highly increased risk (12-15). According to our experience criteria summarized in table 2 are of a particular value in identification of cases suspected of HNPCC.
Table 2. Diagnostic criteria of "suspected HNPCC” (16).
| 1. | among I° relatives of CRC patients (or in himself) at least one cancer of the CRC, endometrium, small bowel or urinary tract |
| 2. | at least one of above cancers diagnosed under age of 50 yrs |
| 3. | FAP is excluded* |
*see table 1
Examples of families matching criteria of "suspected HNPCC” are presented on figures 2-3.
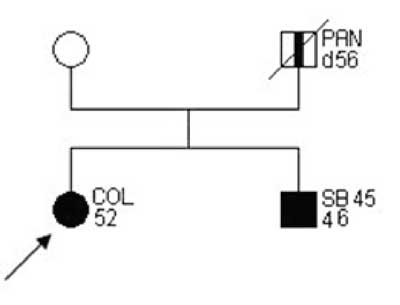
Fig. 2. Family pedigree "suspected HNPCC”.

Fig. 3. Family pedigree "suspected HNPCC”.
Molecular diagnostics of constitutional mutations in genes associated with HNPCC
DNA testing is recommended in families fulfilling at least "suspected HNPCC” criteria. After exclusion of FAP (characteristic FAP features include polyposis, congenital hypertrophy of the retinal pigment epithelium, cysts and osteomata of bones of the maxilla and mondible desmoid tumors) immunohistochemical analyses (IHC) of MLH1, MSH2, MSH6expression in malignant tissues should be performed (absence of the protein may indicate the mutated gene).
Results of several studies performed in our center characterised the frequencies and spectrum of MSH2 and MLH1 mutations in Poland (16). Similary to other populations, the most frequent causes of HNPCC in Poland are MLH1 and MSH2 mutations, constituting 90% of all mutations associated with this syndrome. MLPA detects 10% of these mutations. In over 60% of all HNPCC families recurrent mutations can be found. Thus, after IHC MLPA, for MSH2 and MLH1 should be performed. Next, with MLPA negative, DNA tests searching for recurrent mutations, characteristic for Polish population, should be applied. Last step should include DHPLC (17) and sequencing of the cases indicated by DHPLC results.
Another promising method is the designer iPLEX/TaqMan test plexes, which comprised seven mutations of the APC gene and 29 mutations of three of the mismatch repair genes andseems to be an an outstanding tool for identification of recurrent mutations among hereditary colorectal cancer patients, including HNPCC. (18).
Detection of marker mutation for family with HNPCC is of clinical importance because: A. allows exclusion around 50% relatives from high risk group, B. facilitates decision about surgery extension, for example instead of classical – tumour resection the colectomy with prophylactic hysterectomy and ovariectomy when such resection is performed in diagnosed carriers of mutation – females at perimenopausal age.
Management of families with HNPCC
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Lovett E: Family studies in cancer of the colon and rectum. Br J Surg 1976; 63: 13-18.
2. Lynch HT, Lynch J, Lynch P: Management and control of familial cancer. In: Mulvill JJ, Miller RW, Fraumeni JF, eds. Genetics of Human Cancer. New York, Raven Press 1977; 3: 235-55.
3. Ponz de Leon M, Sassatelli R, Sacchetti C et al.: Familial aggregation of tumors in the three-year experience of a Population-based Colorectal Cancer Registry. Cancer Research 1989; 49: 4344-8.
4. Lynch HT, Smyrk T, Watson P et al.: Hereditary colorectal cancer . Seminars in Oncology 1991; 18: 337-66.
5. Vasen H: Inherited forms of colorectal, breast, and ovarian cancer. Surgical Oncology Clin. N-Am. 1994; 3: 501.
6. Lynch HT, Krush AJ: Cancer family „G” revisited: 1895-1970. Cancer 1971; 27: 1505-11.
7. Fishel R, Lescoe MK, Rao MRS et al.: The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993; 75: 1027-38.
8. Leach FS, Nicolaides NC, Papadopoulos N et al.: Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993; 75: 1215-25.
9. Nicolaides NC, Papadopoulos N, Liu B et al.: Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994; 371: 75-80.
10. Papadopoulos N, Nicolaides NC, Wei Y-F et al.: Mutation of a mutL homolog in hereditary colon cancer. Science 1994; 263: 1625-9.
11. Vasen HF, Mecklin JP, Khan PM, Lynch HT: The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991; 34: 424-5.
12. Vasen HFA, Watson P, Mecklin JP, Lynch H: New Clinical Criteria for Hereditary Nonpolyposis Colorectal Cancer (HNPCC, Lynch syndrome) Proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999; 116: 1453-6.
13. Rodriques-Bigas MA, Boland CR, Hamilton SR et al.: A National Cancer Institute Workshop on Hereditary on-polyposis Colorectal Cancer Syndrome: meeteng highlights and Bethesda Guidelines. J. Nat Cancer Ist 1997; 89: 1758-62.
14. Park JG, Vasen FA, Park KJ et al.: Suspected Hereditary Nonpolyposis Colorectal Cancer. Dis Colon Rectum 1999; 42: 710-6.
15. Park JG, Vasen FA, Park KJ et al.: Suspected HNPCC and Amsterdam criteria II: evaluation of mutation detection rate, an international collaborative study. Int J Colorectal Dis 2002; 17: 109-14.
16. Kurzawski G, Suchy J, Lener M et al.: Germline MSH2and MLH1mutational spectrum including large rearrangements in HNPCC families from Poland (update study). Clin Genet 2006; 69: 40-7.
17. Kurzawski G, Safranow K, Suchy J et al.: Mutation analysis of MLH1and MSH2genes performed by denaturing high-performance liquid chromatography. J Biochem Biophys Meth 2002; 51: 89-100.
18. Dymerska D, Serrano-Fernández P, Suchy J et al.: Combined iPLEX and TaqMan assays to screen for 45 common mutations in Lynch syndrome and FAP patients. J Mol Diagn 2010; 12 (1): 82-90.
19. Vasen HF, Mecklin JP, Watson P et al.: Surveillance in Hereditary Nonpolyposis Colorectal Cancer: an international cooperative study of 165 families.The International Collaborative Group on HNPCC. Dis Colon Rectum 1993; 36: 1-4.
20. Lynch H, Lynch J: Lynch syndrome: Natural history, Genetic Counseling and Prevention. J Clin Oncol 2000; 18: 19-31.
21. Vasen HFA, Möslein G, Alonso A et al.: Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 2007; 44: 353-362.
22. Willet W, Stampfer M, Colditz G et al.: Relation of meat, fat, and fiber intake to the risk of colon cancer in a prospective study among women. N Engl J Med 1990; 323: 1664-72.
23. Burn J, Bishop DT, Mecklin JP et al.: Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndromeN Engl J Med 2008; 359 (24): 2567-78.
24. Bralow SP: Primary and secondary chemoprevention of colorectal cancer: Hereditary colorectal cancer. Springer Verlag Tokyo 1990; 231.
25. Muscat JE, Stellman SD, Wynder EL: Nonsteroidal antiiflamatory drugs and colorectal cancer. Cancer 1994; 74: 1847.
26. Vasen HF, Nagengast FM, Khan PM: Interval cancers in hereditary non-polyposis colorectal cancer (Lynch syndrome). Lancet 1995; 345: 1183-4.
27. Watson P, Vasen HF, Mecklin JP et al.: The risk of endometrial cancer in hereditary nonpolyposis colorectal cancer. Am J Med 1994; 96: 516-20.
28. Järvinen HJ, Aarnio M, Mustonen H et al.: Controlled 15-year Trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology 2000; 118: 829-34.
