© Borgis - Postępy Nauk Medycznych 7/2010, s. 571-576
Joanna Malinowska, Joanna Kołodziejczyk, *Beata Olas
Prophylaxis and treatment of hyperhomocysteinemia
Profilaktyka i leczenie hiperhomocysteinemii
Department of General Biochemistry, University of Łódź
Head of Department: prof. zw. dr hab. Barbara Wachowicz
Streszczenie
Homocysteina (Hcys) jest niebiałkowym aminokwasem będącym metabolitem pośrednim na szlakach przemiany metioniny. Podwyższone stężenie homocysteiny (> 15 ?M, tzw. hyperhomocysteinemia) jest skorelowane z występowaniem u ludzi schorzeń, takich jak choroby sercowo-naczyniowe, choroby neurodegeneracyjne oraz choroby nerek. Wyróżnia się hiperhomocysteinemię łagodną, umiarkowaną i ciężką, gdzie stężenie Hcys w osoczu mieści się w przedziałach: 16-30 ?M, 31-100 ?M oraz przekracza 100 ?M. Prawidłowe całkowite stężenie Hcys w osoczu mieści się w przedziale od 5 do 15 ?M (średnio 10 ?M). Homocysteina występuje we krwi człowieka w kilku formach, jako forma niezwiązana (np. jak forma zredukowana w stężeniu 100 nM) oraz w połączeniu z białkami, ale najbardziej aktywną formą homocysteiny jest tiolakton homocysteiny (HTL). W niniejszej pracy przedstawiono kilka dróg metabolizmu Hcys, który jest skorelowany z poziomem różnych witamin (witaminy B6, B12 i kwasu foliowego). Ponadto, genetyczne i środowiskowe przyczyny hiperhomocysteinemii oraz profilaktyka i leczenie hiperhomocysteinemii zostały także omówione.
Summary
Homocysteine (Hcys) is a non-protein sulfur-containing amino acid, an intermediate product of methionine metabolism. It is known that elevated concentration of homocysteine (> 15 ?M; hyperhomocysteinemia) in humans is correlated with various diseases, including cardiovascular, neurodegenerative and kidney disorders. Hyperhomocysteinemia has been classified into three types, depending on the plasma levels of Hcys – mild (16-30 ?M), moderate (31-100 ?M) and severe (> 100 ?M). Normal plasma level of homocysteine ranges from 5 to 15 ?M (mean 10 ?M). Homocysteine exists in the blood in several forms: in free (e.g. as reduced form – about 100 nM) and protein bound form, but the most reactive form of Hcys is homocysteine thiolactone (HTL). In the article, some pathways of Hcys metabolism, which is correlated with the level of various vitamins (B6, B12 and folic acid), are described. Moreover, genetic and environmental causes of hyperhomocysteinemia, as well as prophylaxis and treatment of hyperhomocysteinemia, are also discussed.

INTRODUCTION
Homocysteine (Hcys) is a homologue of the amino acid cysteine, at the same time being a derivate of methionine, from which it differs by the lack of a methylene group at the sulfur atom (fig. 1). Hcys was discovered in the 1930s, together with cysteine and glutathione. These three compounds constitute a group of low-molecular-mass biological thiol compounds. Elevated plasma level of homocysteine is considered to be an unequivocal risk factor of cardiovascular diseases. There are many literature reports available showing elevated Hcys level as an independent factor of premature atherosclerosis development (besides hypercholesterolemia). The role of Hcys in pathogenesis of vascular disorders, as well as its procoagulant effect, results from its influence on blood vessel walls, blood platelets (fig. 2) and the coagulation/fibrinolysis proteins. Detailed descriptions of the effect of Hcys on selected hemostasis elements, including blood platelets and coagulation, have been provided by Karolczak and Olas (1), as well as by Malinowska et al. (1). Elevated homocysteine level (hyperhomocysteinemia) in blood can result from genetic, congenital metabolic anomalies or from environmental factors (acquired, in connection with lifestyle). It is estimated that every one in ten Europeans has an elevated blood Hcys level, which leads to increased cardiovascular risk, including atherosclerosis and myocardial infarction.
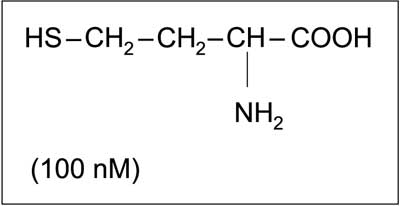
Fig. 1. Chemical structure of homocysteine in reduced form and its concentration in human blood (1, modified).
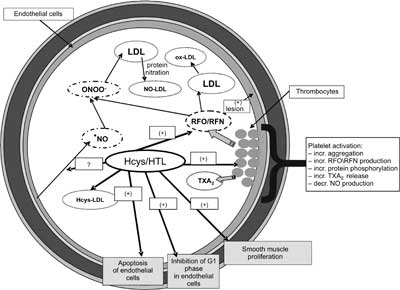
Fig. 2. Regulatory role of homocysteine and its thiolactone in blood vessel wall and blood platelets. LDL – low density lipoproteins, Hcys-LDL – homocysteinylated LDL, ox-LDL – oxidized LDL, RFN – reactive forms of nitrogen, RFO – reactive forms of oxygen, NO?-nitric oxide, NO-LDL – nitrated LDL, TXA2 – thromboxane A2 (1, modified).
Moreover, increased blood Hcys levels are observed in patients with chronic renal failure, hypothyroidism, various cancer types, malignant anemia, liver diseases, and in persons with malnutrition, especially with low folic acid intake. It should also be noted that an increase in Hcys levels correlates with the severity of some other disorders, including Alzheimer's disease and other types of dementia, Parkinson's disease, multiple sclerosis, and chronic fatigue syndrome.
METABOLISM OF HOMOCYSTEINE AND ITS THIOLACTONE
Proteins that are absorbed with food are metabolized in the human body into amino acids. One of the products of protein degradation is the amino acid methionine (Met). Some of the Met is recycled and used again for protein synthesis, while the remaining portion may be metabolized into homocysteine (2). Hcys is a thiol amino acid, which is synthesized in all types of animal (including human) cells. S-adenosylmethionine transferase (SAM synthetase) catalyses the reaction of adenosine transfer (from ATP) to methionine. S-adenosylmethionine (SAM) is the main donor of the methyl group (–CH3) for various methylation reactions. After the methyl group is transferred by SAM synthetase to various compounds, SAM turns into S-adenosylhomocysteine (SAH). Subsequently SAH is hydrolyzed by specific SAH hydrolase into adenosine and homocysteine (fig. 3) (3), and its plasma level can be measured. Moreover, it is known that Hcys does not have its own codon, so it cannot be incorporated into protein chains in the way that other amino acids can (4). But it can bind with them through the ε-amino group of lysine. Hcys can also bind to proteins and/or cysteine via disulfide bonds. It has been shown that approximately 33% of the total Hcys circulates as oxidized low-mass disulphide compounds, 1% as a free, reduced form, and about 66% is bound to proteins (as N-homocysteinylated – or S-homocysteinylated proteins) (5, 6).
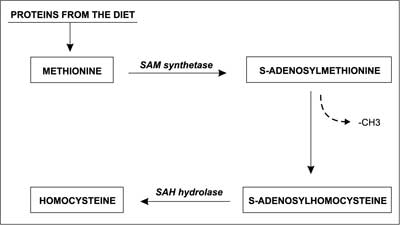
Fig. 3. Formation of homocysteine from methionine (4, modified).
Literature reports describe 2 pathways of Hcys metabolism. The first one is based on re-methylation of Hcys into methionine, with substantial involvement of folic acid, which acts here as a methyl group donor (7, 8). Vitamin B12 (cobalamin) and Vitamin PP (niacin) are necessary co-factors for metabolism of tetrahydrofolate derivates (9). The second pathway leads to cysteine through Hcys transsulfuration, with pyridoxal-5-phosphate as a co-factor (active form of vitamin B6) (fig. 4) (10, 11).
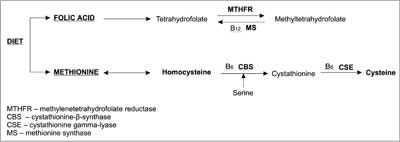
Fig. 4. Homocysteine metabolism and the role of vitamins B6, B12 and folic acid in Hcys transformations (11, modified).
Another form of Hcys in plasma is its thiolactone, homocysteine thiolactone (HTL) (fig. 5). HTL is much more reactive than Hcys. Its synthesis results from erroneous homocysteine activation, e.g. by methionyl-t-RNA synthetase (MetRS) (12). It is worth noting that homocysteine thiolactone may undergo non-enzymatic hydrolysis, but mostly it is metabolized by plasma HTL hydrolase, also known as homocysteine thiolactonase (HTLase) or paraoxonase (PON 1).
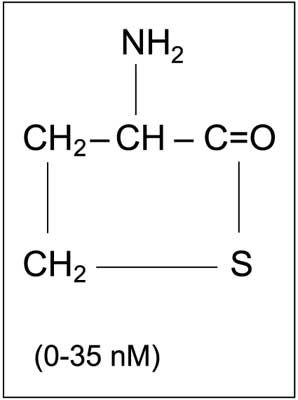
Fig. 5. Chemical structure of homocysteine thiolactone and its concentration in human blood (1, modified).
It has been observed that impaired metabolism of homocysteine leads to a rise of its level in the blood and further to various abnormalities. Normal blood Hcys levels range from 5 to 15 μM. A preprandial level of Hcys exceeding 15 μM is termed hyperhomocysteinemia. There are three types of this disorder – mild, moderate and severe – with plasma Hcys levels 16-30 μM, 31-100 μM and above 100 μM, respectively. Although the severe form is rare, 5-7% of the general population suffer from mild hyperhomocysteinemia. Subjects with mild form of hyperhomocysteinemia do not show clinical symptoms until they reach 40 years of age, when signs of premature atherosclerosis start to show. Moderate hyperhomocysteinemia can pose a significant danger for young women, as it increases the risk of complications during pregnancy (13).
GENETIC FACTORS IN HYPERHOMOCYSTEINEMIA
In recent years numerous data on possible genetic causes of cerebrovascular diseases have been collected. It has been observed that stroke occurs significantly more often in twins, especially monozygotic twins, as well as in subjects whose parents suffered from cerebral stroke (14). A significant clinical role here has been attributed to genetically conditioned alterations of homocysteine metabolism.
An elevated Hcys level may result from genetic defects of enzymes involved in its metabolism, among others cystathionine-β-synthase (CBS), methionine synthase (MS) and methylenetetrahydrofolate reductase (MTHFR). Heterozygous lack of CBS, CBS mutations and methylenetetrahydrofolate reductase gene polymorphism are thought to be the most probable causes of hyperhomocysteinemia. In CBS, Ile278Thr and Gly307Ser are the most common mutations. If they occur homozygously, they may lead to significant elevation of homocysteine level and to an increased risk of atherosclerosis of carotid arteries (15). Methylenetetrahydrofolate reductase gene polymorphism involves substitution of cytosine by thymine in position 677 (C677&ro;T). This nucleotide exchange leads to the synthesis of a thermolabile enzyme form with decreased activity. As a result synthesis of methylenetetrahydrofolate (donor of methyl group in re-methylation of homocysteine) is decreased. Prevalence of this mutation is race dependent. In Asian and Caucasian populations (including Poland) there are 50% C7T heterozygotes and 10-13% T/T homozygotes, while in the Afro-American population cases of this polymorphism are rare (16). T/T homozygotes have on average 2.5 μM higher levels of Hcys than C/C homozygotes. However, this difference has been observed in the case of folic acid and riboflavin deficiency in the diet (17). It has also been observed that A&ro;C transversion in position 1298 of the MTHFR gene leads to substitution of glutamate with alanine in the enzyme, which leads to its decreased activity (18).
ENVIRONMENTAL FACTORS IN HYPERHOMOCYSTEINEMIA
Acquired hyperhomocysteinemia may result from various factors. Among others, lifestyle, diet, various diseases, medications, drugs, sex and age influence levels of Hcys. Dietary errors leading to deficiency of vitamins B2, B6, B12 and folic acid may in turn cause an increase of Hcys level. Also alimentary tract disorders connected with impaired absorption of these vitamins may play an important role.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Malinowska J, Nowak P, Olas B: Hiperhomocysteinemia, a zaburzenia procesu hemostazy – fakty i mity. Polski Merkuriusz Lekarski 2009; 27: 5-10.
2. Bełtowski J: Protein homocysteinylation: a new machanism of atherogenesis? Postępy Hig Med Dosw 2005; 59: 392-404.
3. Mitchell T: Methylation a little-know but essential process. Life Extension 1998; 09: www.lef.org/magazine/mag/aug98-report2.html.
4. Durand P, Prost M, Blache D: Pro-thrombotic effects of a folic acid deficient diet in rat platelets and macrophages related to elevated homocysteine and decreased n-3 polyunsaturated fatty acids. Atherosclerosis 1996; 121: 231-43.
5. Bathum L, Petersen I, Christiansen L et al.: Genetic and environmenatal influences on plasma homocysteine: Results from a Danish twin study. Clin Chem 2007; 53: 971-9.
6. Bogdański P, Pupek-Musialik D, Łuczak M et al.: Ocena stężenia homocysteiny i wybranych markerów procesu zapalnego u chorych z klinicznymi cechami insulinooporności. Diabet Dosw Klin 2003; 3: 261-7.
7. Medina M, Urdiales JL, Amores-Sanchez MI: Roles of homocysteine in cell metabolism: old and new functions. Eur J Biochem. 2001; 268: 3871-82.
8. Perła-Kaján J, Twardowski T, Jakubowski H: Mechanism of homocysteine toxicity in humans. Amino Acids 2007; 32: 561-72.
9. Undas A, Stepien E, Plicner D et al.: Elevated total homocysteine is associated with increased platelet activation at the site of microvascular injury. Effects of folic acid administration. J Thromb Haemost 2007; 5: 1070-2.
10. Banecka-Majkutewicz Z, Gąsecki D, Jakóbkiewicz-Banecka J et al.: Hiperhomocysteinemia – ważny czynnik ryzyka udaru mózgu. Hyperhomocysteinemia- important risk factor for ischemic stroke. Udar Mózgu 2005; 7, 2: 61-5.
11. Kraczkowska S, Suchocka Z, Pachecka J: Podwyższone stężenie homocysteiny we krwi jako wskaźnik zagrożenia zdrowia. Biul Wydz Farm AMW 2005; 3.
12. Bromme D, Rossi AB, Smeekens SP et al.: Human bleomycin hydrolase: molecular cloning, sequencing, functional expression, and enzymatic characterization. Biochemistry 1996; 35: 6706-14.
13. Graham IM, Daly LE, Refsum HM et al.: Plasma homocysteine as a risk factor for vascular disease. JAMA 1997; 277: 1775-81.
14. Hankey G, Eikelboom J: Homocysteine and vascular disease. Lancet 1999; 354: 407-13.
15. Klerk M, Verhoef P, Clarke R et al.: MTHFR 677C&ro;T polymorphism and risk of coronary heart disease: meta-analysis. JAMA 2002; 288: 2023-31.
16. Brattstörm L, Wilcken DE, Ohrvik J, Brudin L: Common methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: The result of a meta-analysis. Circulation 1998; 98: 2520-6.
17. Bednarek-Tupikowska G, Tupikowski K: Homocysteine- an underestimated atheromatosis risk factor. Do sex hormones influence homocysteine concentrations? Homocysteina – niedoceniany czynnik ryzyka miażdżycy. Czy hormony płciowe wpływają na stężenie homocysteiny? Postepy Hig Med Dosw 2004; 58: 381-9.
18. Chango A, Boisson F, Barbe F et al.: The effect of 677 C(r)T and 1298 A(r)C mutations on plasma homocysteine and 5,10-methylenetetrahydrofolate reductase activity in healthy subjects. Br J Nutr 2000; 83: 593-6.
19. Rousseau AS, Robin S, Roussel AM et al.: Plasma homocysteine is related to folate intake but not training status. Nutrition, Metabolism and Cardiovascular Diseases 2005; 15: 125-33.
20. Gąsiorowska D, Korzeniowska K, Jabłecka A: Homocysteine. Farm Współ 2008; 1: 169-75.
21. Piechota W, Piechota W: Correlation of homocysteine concentration with the extent of coronary ahterosclerosis in men with symptoms of ischemic heart disease. Korelacja stężeń homocysteiny ze zmianami w tętnicach wieńcowych u mężczyzn z objawami choroby niedokrwiennej serca. Pol Przegl Kardiol 2004; 6: 401-6.
22. Domagała B, Sanak M, Czachór R, Szczeklik A: Hiperhomocysteinemia i jej związek z miażdżycą tętnic. Pol Arch Med Wew 1997; 98: 153-62.
23. Supiński W, Głuszek J, Pawlak A, Srauss E: Czy obniżenie homocysteiny po podaniu vitamin z grupy B może zmniejszać liczbę powikłań sercowo-naczyniowych? Czynniki Ryzyka 2007; 2: 29-37.
24. Kaul S, Zadeh AA, Shah PK: Homocysteine hypothesis for atherothrombotic cardiovascular diseases. J Am Coll Cardiol 2006; 48: 914-23.
25. Kozłowska Wojciechowska M: Jak zapobiegać hiperhomocysteinemii? Naturalne źródła folianów i vitamin z grupy B w polskiej diecie. Czyn Ryzyka 2005; 11: 25-6.
26. Bald E: Homocysteina, niegdyś egzotyczny metabolit. [W:] Biotiole w warunkach fizjologicznych, patologicznych i w terapii. Wydawnictwo Uniwersytetu Jagiellońskiego 2003; 73-108.
27. Zalecenia Polskiego Towarzystwa Badań Nad Miażdżycą – http://www.dach-liga-homocystein.org/.
28. Rechciński T: Hiperhomocysteinemia – patomechanizmy, diagnostyka, leczenie. Forum Kardiologów 2001; 6: 67-9.
