Aktywność farmakologiczna biflawonoidów. Część I
The pharmacological activity of biflavonoids. Part I
Katedra i Zakład Farmakognozji z Ogrodem Roślin Leczniczych, Akademia Medyczna w Gdańsku
Kierownik: prof. dr hab. Wojciech Cisowski
Biflawonoidy są jedną z unikatowych klas występujących w przyrodzie związków flawonoidowych (9, 17, 19). Pierwszy biflawon – ginkgetyna, został wyizolowany jako żółty barwnik z liści Ginkgo biloba przez Furukawa, w 1929 roku (16). Obecnie w literaturze jest opisanych około 200 struktur biflawonoidowych wyodrębnionych z roślin i reprezentujących pochodne około 21 podstawowych układów (17, 19).
Biflawonoidy są dimerycznymi formami aglikonów flawonoidowych – flawonów, flawanonów a także chalkonów i flawonoli (17). W zależności od charakteru wiązania między dwiema jednostkami flawonylowymi biflawonoidy dzielimy na:
I –posiadające wiązanie C-C np. pochodne apigeniny połączone w pozycjach (ryc. 1):
np. pochodne apigeniny połączone w pozycjach:
C6-O-C4´´´– typ hinokiflawonu
C3´-O-C4´´´– typ ochnaflawonu (17).
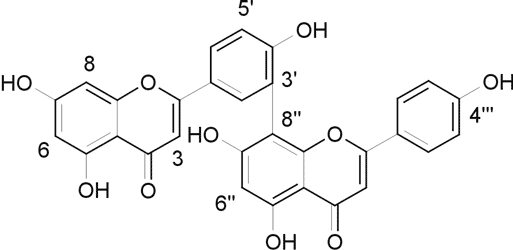
Ryc. 1. Typ amentoflawonu.
W ciągu ostatnich dziesięciu lat intensywnie badano aktywność farmakologiczną biflawonoidów, wcześniej w niewielkim stopniu rozpoznaną (17), szczególnie na tle innych grup połączeń flawonoidowych (19, 32, 37). Niniejsze opracowanie stanowi podsumowanie wyników przeprowadzonych prac, uwzględniając w części pierwszej przeciwzapalne (7, 10, 11, 12, 18, 22-25, 28-30, 35), antyalergiczne (2, 8), przeciwbólowe (6, 31, 38), antyhepatotoksyczne (3, 14, 21, 34) i przeciwcukrzycowe (15, 38) właściwości dimerów flawonoidowych.
Wiele substancji chemicznych z grupy flawonoidów wykazuje w warunkach in vitro i in vivo działanie przeciwzapalne, jednak siła tej aktywności dla przeprowadzenia badań klinicznych jest niewystarczająca (32). Intensywne poszukiwania nowych flawonoidowych molekuł w perspektywie ich zastosowania jako leków przeciwzapalnych są kontynuowane (7, 10-12, 18, 22-25, 28-30, 35).
W oparciu o badania na zwierzęcych modelach ostrego i przewlekłego odczynu zapalnego, dwa dimery flawonowe – amentoflawon i ginkgetynę, w porównaniu do niesteroidowych i steroidowych leków przeciwzapalnych, uznano za związki o aktywności przeciwzapalnej (22-24, 28, 30). Potwierdzono, że mechanizm ich działania jest związany z bezpośrednim hamowaniem enzymów metabolizujących kwas arachidonowy, oraz z supresją ekspresji enzymów odczynu zapalnego (22, 28).
Działanie przeciwzapalne ochnaflawonu i ginkgetyny jest wyjaśniane ich właściwościami hamowania fosfolipazy A2 (PLA2) (11, 22, 25, 30), biorącej udział w uwalnianiu arachidonianów z fosfolipidów błon komórkowych (27). Kwas arachidonowy stanowi substrat dla lipooksygenazy (LOX) i cyklooksygenazy (COX), enzymów biosyntezy leukotrienów, prostaglandyn i tromboksanów. Eikozanoidy, obok innych endogennych substancji, są mediatorami zapalenia, bólu i gorączki, dlatego zahamowanie ich biosyntezy wiąże się z działaniem przeciwzapalnym, przeciwbólowym, przeciwgorączkowym a także przeciwalergicznym. Między innymi leukotrieny LTC4 i LTD4 silnie kurczą naczynia krwionośne i mięśnie gładkie oskrzeli oraz zwiększają przepuszczalność naczyń włosowatych, natomiast prostacyklina PGI2 hamuje agregację płytek krwi oraz rozszerza naczynia krwionośne(27).
Ginkgetynę, izoginkgetynę, amentoflawon, apigeninę i staurosporynę poddano eksperymentowi, którego celem było określenie zdolności hamowania uwalniania PLA2 z makrofagów (30). W przypadku gdy aktywatorem uwalniania PLA2 był PMA (12-mirystyniano-13-octan forbolu), dwa pierwsze związki w stężeniu 10µM, wykazywały skuteczność 32,5% i 40%, odpowiednio. Natomiast gdy jako aktywator zastosowano jonofor A23187 (związek warunkujący transport jonów wapniowych przez błony komórkowe) (27), skuteczność ginkgetyny wynosiła 21,7%, izoginkgetyny 41,7%, przy jednoczesnym braku aktywności amentoflawonu. Amentoflawon posiadał działanie ochronne jedynie w stosunku do indukowanego PMA uwalniania kwasu arachidonowego. Apigenina nie charakteryzowała się znaczącym wpływem na PLA2, natomiast staurosporyna – inhibitor proteinowej kinazy C, posiadała wyraźne cechy inhibitora PLA2, który już w stężeniu 1µM wykazywał 96,8% skuteczność (30).
Ochnaflawon izolowany z Lonicera japonica (Ochnaceae) silnie inaktywował płytkową fosfolipazę A2 (IC50=3µM) (11). Stopień inaktywacji zależał od wartości pH, przy czym maksymalny efekt obserwowano w zakresie pH 9-10. Ujawniono większą specyficzność ochnaflawonu w stosunku do grupy II fosfolipazy A2 niż do grupy I (IC50=20µM). Pomimo że PLA2 jest aktywowana przez wapń i kalmodulinę (27), dodatek nadmiaru jonów Ca+2 (stężenie powyżej 8µM) nie antagonizował aktywności ochnaflawonu (11).
Odnotowano w porównaniu do słabo aktywnej kwercetyny, znaczące hamowanie epidermalnej fosfolipazy A przez amentoflawon i ginkgetynę, potwierdzając pogląd, że poprzez modulację aktywności tego enzymu flawonoidy mogą ujawniać zróżnicowane efekty w metabolizmie kwasu arachidonowego (25).
Morelloflawon in vitro ograniczał sekrecję PLA2 wywołaną m.in. enzymami jadów pszczół (IC50=0,6µM), natomiast w stosunku do cytozolowej PLA2 z ludzkich monocytów był nieaktywny (18). Jednocześnie, związek ten na modelach zwierzęcych zmniejszał obrzęk wywołany 13-octanem-12-O-tetradekanoiloforbolu (TPA) i karageniną, nie zmieniając poziomu eikozanoidów w homogenatach tkanek objętych zapaleniem. W procesach uwalniania reaktywnego tlenu przez neutrofile, aktywowanym luminolem i lucygeniną, związek wykazywał wartości IC50 odpowiednio 2,7µM i 1,8 µM. Ustalono, że morelloflawon jest selektywnym inhibitorem PLA2, szczególnie grupy II i III enzymów. Założono, że jego przeciwzapalne działanie wynika z właściwości zmiatania wolnych rodników, a nie z bezpośredniego wpływu na kaskadę kwasu arachidonowego (18). Hydroksynadtlenki powstające m.in. podczas przemiany kwasu arachidonowego, w tak zwanym sprzężeniu zwrotnym pozytywnym, za pośrednictwem wolnych rodników nasilają aktywność COX. W ten sposób zmiatacze wolnych rodników mogą osłabiać aktywność COX (27). Powyższy mechanizm działania być może charakteryzuje morelloflawon.
Porównano wpływ pięciu typów połączeń flawonoidowych – flawanonu, flawonu-apigeniny, flawonolu-kwercetyny, dimeru flawonowego-amentoflawonu na aktywność epidermalnej cyklooksygenazy (COX) i lipooksygenazy (LOX). Za źródło COX i LOX przyjęto mikrosomalne i cytozolowe frakcje homogenatu naskórka świnek morskich. Amentoflawon zaznaczył się silnym i selektywnym działaniem wobec COX (IC50=3µM), zbliżonym do indometacyny (IC50=1µM). Natomiast ginkgetyna – dimetylowa pochodna amentoflawonu była słabym inhibitorem COX. Na tle kwercetyny silnie hamującej obydwa enzymy, flawanon i apigenina były nieaktywne (26).
Amentoflawon izolowany z korzeni Biophytum sensitivum (Oxalidaceae) w teście in vitro okazał się selektywnym inhibitorem cyklooksygenazy-1 (oznaczona wartość IC50=12,4µM w porównaniu do IC50=1,1µM indometacyny jako kontroli). Związek przy wyższych wartościach IC50>37µM nieznacznie hamował aktywność cyklooksygenazy-2, enzymu uwalnianego w procesach zapalnych (10).
Ginkgetyna otrzymana z liści Ginkgo biloba, ograniczała syntezę prostaglandyny PGE2 (65,6% zahamowania) oraz wpływała na poziom cyklooksygenazy-2 poprzez supresję indukcji tego enzymu. Ponadto związek hamował in vivo, po podaniu miejscowym (100-1000 mg), obrzęk ucha myszy w modelu zapalenia indukowanym olejem krotonowym (28).
Dwa flawano-flawanonowe glikozydy – dyinzyninol i dyinzynę izolowano jako związki o aktywności przeciwzapalnej z korzeni Sarcophyte piriei (Balanophoraceae). W przeprowadzonych testach, hamowały one syntezę prostaglandyn (IC50=9,20µM i 13,14µM, odpowiednio) oraz egzocytozę indukowaną PAF (Plateled Activated Factor) (IC50 =49µM i 39µM, odpowiednio). Powyższe związki zostały wyodrębnione z ekstraktu wodnego Sarcophyte piriei, którego działanie przeciwzapalne w warunkach in vitro wyrażone mocą zdolności hamowania syntezy prostaglandyn było identyczne z aspiryną (IC50=0,2 µM) (35). Badany gatunek jest rośliną pasożytniczą, porastającą korzenie różnych gatunków akacji rosnących w Afryce. Odwar z podziemnych bulw jest popularnym przeciwzapalnym lekiem roślinnym, stosowanym w stłuczeniach, bólach gardła, zębów i brzucha w medycynie ludowej Somalii (35).
Flawony i flawonole ujawniają in vitro i in vivo działanie przeciwzapalne, które jest nie tylko związane z ich właściwościami antyutleniającymi, wpływem na metabolizm kwasu arachidonowego (22, 32, 33), ale również z hamowaniem powstawania tlenku azotu (NO), odgrywającego istotną rolę w patologii zapaleń via aktywne w stanie zapalnym syntazy NO (iNOS – NOS typu 2) (33). Wyniki badań aktywności biflawonoidów – ginkgetyny i bilobetyny, obok prenylowanych flawonoidów moruzyny, kuwanonu C, sangenonu D, ujawniły, że wszystkie substancje zmniejszały stymulowaną lipopolisacharydem (LPS) produkcję NO w makrofagach linii komórkowej – RAW 264.7. Efekt ten był spowodowany osłabieniem indukcji enzymu iNOS, a nie bezpośrednim hamowaniem jego aktywności. Wyjątek w analizowanej grupie związków stanowił echinoizoflawon, który obok supresji indukcji iNOS hamował bezpośrednio ten enzym (IC50=83µM). W kontraście do biflawonów, większość prenylowanych pochodnych charakteryzowała się cytotoksycznością wobec komórek RAW w stężeniach 10-100 µM (12). Wykazanie hamowania produkcji NO przez dimery flawonowe, obok inhibicji uwalniania kwasu arachidonowego, częściowo tłumaczy ich immunoregulacyjne i przeciwzapalne działanie in vivo (12).
Kolejnym doniesieniem o możliwym mechanizmie przeciwzapalnej aktywności flawonoidów są wyniki prac opublikowane przez Lee i wsp. (29). Badali oni efekt biflawonoidów na proliferację limfocytów w warunkach in vitro. Z grupy 9 dimerów objętych eksperymentem pięć w stężeniu 10 µM hamowało proliferację indukowaną przez konkanawalinę A (Con A) oraz lipopolisacharyd (LPS) – ochnaflawon, ginkgetyna, izoginkgetyna, kryptomeryna B, izokryptomeryna. Najsilniejsze działanie, w stężeniu 1 µM, posiadały dwa ostatnie związki. Bilobetyna (10 µM) była aktywna jedynie w modelu z LPS-indukowaną proliferacją. Ponadto ochnaflawon i kryptomeryna posiadały podobne profile supresji w stosunku do kultur mieszanych limfocytów. Interesującym jest fakt, że flawony i flawonole w porównaniu do nieselektywnych wobec limfocytów biflawonów, oddziaływały głównie z limfocytami T. W oparciu o wyniki przeprowadzonych badań oraz uwzględniając wcześniejsze dane piśmiennictwa o wpływie biflawonoidów na PLA2, wskazano na ochnaflawon jako potencjalny lek przeciwzapalny w leczeniu chorób o podłożu immunologicznym, takich chociażby jak reumatoidalne zapalenie stawów (29).
Frakcja dimerów flawonoidowych otrzymana z nasion Garcinia kola (Guttiferae) nazwana kolawironem w warunkach in vivo wykazywała skuteczne działanie przeciwzapalne, zmniejszając, w dawce 100 mg/kg, indukowane karageniną, obrzęk łapy i turpentyną obrzęk stawu szczura. Odnotowano również działanie przeciwgorączkowe kolawironu. Stwierdzono, że siła aktywności przeciwzapalnej kolawironu jest porównywalna z fenylobutazonem i kwasem acetylosalicylowym, zastosowanym jako przeciwzapalne związki standardowe (7).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Adaramoye O.A., Akinloye O.: Biosci. Rep. 2000, 20:259. 2.Amellal M. et al.: Planta Med. 1985, 51:16. 3. Anand K.K. etal.: Planta Med. 1992, 58:493. 4. Beretz A. et al.: Biochem. Pharmacol. 1986, 35:257. 5. Bin J.I. et al.: Pharm Biol. 2001, 39:243. 6.Bittar M. et al.: Planta Med. 2000, 66:84. 7. Braide V.B.: Fitoterapia 1993, 64:433. 8. Bronner C., Landry Y.: Agents Actions 1985, 16:147. 9. Bruneton J.: Pharmacognosy. Intercept, Paris 2001. 10. Bucar F. et al.: Planta Med. 1997, 64:373. 11. Chang H.W. et al.: Biochem. Biophys. Res. Commun. 1994, 205:843. 12.Cheon B.S. et al.: Planta Med. 2000, 66:596. 13.Cholbi M.R. et al.: Experientia 1991, 47:195. 14. Farombi E.O.: Pharmacol. Res. 2000, 42:75. 15. Felicio J.D. et al.: Planta Med. 1995, 61:217. 16. Furukawa S.: Sci. Papers Inst. Phys. Chem. Research Tokyo 1933, 21:273. Cyt. wg Chem. Abstr. 1933, 27:5745. 17. Geiger H., Quinn Ch.: Biflavonoids. In: The Flavonoids. Advances in Research (red. J.B. Harborne). Chapman and Hall, London, 1988, 111. 18. Gil B. et al.: Biochem. Pharmacol. 1997, 53:733. 19. Harborne J.B.: The Flavonoids. Advances in Research. Chapman and Hall, London 1988. 20.Iwu M.M. et al.: J. Pharm. Pharmacol. 1990, 42:290. 21.Iwu M.M. et al.: J. Ethnopharmacol. 1987, 21:127. 22. Kim H.K. et al.: Nat. Prod. Sci. 2000, 6:170. 23. Kim H.K. et al.: Arch. Pharm. Res. 1998, 21:406. 24. Kim H.K. et al.: Planta Med. 1999, 65:465. 25. Kim H.P. et al.: Fatty Acids 1998, 58:17. 26.Kim H.P. et al.: Prostaglandins Leukotrienes Essent. Fatty Acids 2001, 65:281. 27. Kostowski W.: Farmakologia. PZWL, Warszawa 2001. 28. Kwak W.J. et al.: Planta Med. 2002, 68:316. 29. Lee S.J. et al.: Life Sci. 1995, 57:551. 30. Lee S.J. et al.: Archiv. Pharmacol. Res. 1997, 20:533. 31. Luzzi R. et al.: Phytomedicine 1997, 4:141. 32. Middleton E., Kandaswami C.: Biochem. Pharmacol. 1992, 43:1167. 33. Mora A. et al.: Biochem. Pharmacol. 1990, 40:793. 34. Nwankwo J.O. et al.: Eur. J. Cancer Prev. 2000, 9:351. 35. Ogundaini A. et al.: J. Nat. Prod. 1996, 59:587. 36. Okunji C.O., Iwu M.M.: Fitoterapia 1990, 62:74. 37. Pathak D. et al.: Fitoterapia 1991, 62:371. 38. Silva K.L. et al.: Therapie 2001, 12:431.

