© Borgis - Postępy Nauk Medycznych 11/2010, s. 832-841
*Barbara Jarząb, Jan Włoch, Zbigniew Wygoda
Clinical genetics of medullary thyroid carcinoma
Genetyka kliniczna raka rdzeniastego tarczycy
Department Nuclear Medicine and Endocrine Oncology, Maria Skłodowska-Curie Memorial Cancer Center and Institute of Oncology Gliwice Branch, Gliwice, Poland
Head of Department: prof. dr hab. med. Barbara Jarząb
Streszczenie
Rak rdzeniasty tarczycy (RRT) jest neuroendokrynnym nowotworem złośliwym, wywodzącym się z okołopęcherzykowych komórek C. Komórki te pochodzą z grzebienia nerwowego, w czasie rozwoju płodowego migrują z V kieszonki skrzelowej do tarczycy, gdzie produkują kalcytoninę. Kalcytonina jest hormonem peptydowym, ułatwiającym przejście wapnia z krwi do kości.
Rak rdzeniasty tarczycy występuje w postaci sporadycznej oraz dziedzicznej, której wystąpienie związane jest z obecnością mutacji protoonkogenu RET. Dziedzicznemu RRT mogą nie towarzyszyć żadne inne objawy i mówi się wówczas o rodzinnym raku rdzeniastym tarczycy (ang. familial medullary thyroid carcinoma, FMTC). Częściej jednak dziedziczny RRT jest objawem zespołu gruczolakowatości wewnątrzwydzielniczej typu 2 (ang. multiple endocrine neoplasia type 2, MEN 2).
Zespół gruczolakowatości wewnątrzwydzielniczej typu 2A (MEN 2A), zwany również zespołem Sipple'a, charakteryzuje się skojarzeniem RRT z guzami chromochłonnymi nadnerczy (u około 50% chorych) i gruczolakami lub hiperplazją przytarczyc (u około 15-25% chorych). Rozpoznanie zespołu MEN 2B jest daleko bardziej jednoznaczne, tak ze względu na charakterystyczny obraz kliniczny jak i charakterystyczne mutacje. W tym zespole RRT rozwija się najszybciej, jeszcze u małych dzieci. Guzy chromochłonne nadnerczy występują później i ujawniają się u około połowy chorych, natomiast nadczynność przytarczyc nie występuje.
W pracy przedstawiono aktualny stan wiedzy na temat molekularnego podłoża dziedzicznej postaci RRT oraz zależność między lokalizacją mutacji punktowej RET i obrazem klinicznym choroby. Omówiono również postępowanie diagnostyczne i lecznicze w dziedzicznej postaci RRT oraz postępowanie w razie wykrycia nosicielstwa mutacji protoonkogenu RET. Jednocześnie w podsumowaniu podano krótkie wskazówki dotyczące postępowania w przypadku wykrycia dziedzicznej postaci RRT.
Summary
Medullary thyroid carcinoma (MTC) is neuroendocrine malignant neoplasm, arising from the parafollicular thyroid cells. These cells arising from the neural crest, migrating from the fifth branchial cleft into thyroid gland during the embryogenesis, where the calcitonin hormone is producing by them. Calcitonin is the peptide hormone, facilitating calcium transition from the blood to the bones.
Medullary thyroid carcinoma occurs in the sporadic and hereditary form, which presence is connected with proto-oncogene RET mutations. Hereditary form of MTC can be divided into familial medullary thyroid carcinoma (FMTC) without any endocrinopathies and, more frequently, as a part of multiple endocrine neoplasia type 2 (MEN 2).
Multiple endocrine neoplasia type 2A (MEN 2A), named as Sipple syndrome also, can be characterized as presence of MTC and pheochromocytoma (in about 50% of patients) and parathyroid adenomas or hyperplasia (15-25% of patients). Recognition of syndrome MEN2B is more unequivocal because of characteristic clinical status and characteristic mutations. In this syndrome, MTC develops the most quickly, even in young children. Pheochromocytomas occur later and in the half of patients, parathyroid adenomas are absent.
In this paper actual state of knowledge about the molecular basis of hereditary form of MTC and dependence between localization of RET mutations and clinical disease status are presented. Diagnostic and therapeutic procedures in hereditary form of MTC and the way of proceeding in the case of RET mutation presence are discussed Short guidelines about management in the case of hereditary form of MTC are presented also.

Medullary thyroid carcinoma is a malignant neuroendocrine tumour originating within parafollicular cells (C-cells). In the literature its discovery is connected with Hazard (1). However, it should be emphasized that the first reports describing this type of carcinoma were published in the Polish literature, i.e. in "Nowotwory” by Prof. Laskowski who called this type of carcinoma "ca hyalinicum”.
C cells are derived from neural crest cells; they migrate from the fifth pharyngeal pouch to the thyroid where they produce calcitonin. Calcitonin is a peptide hormone facilitating the transfer of calcium from the blood to the bones.
Medullary thyroid carcinoma (MTC) is usually located in the middle and upper part of lateral thyroid where the largest number of parafollicular cells are found. Cancerous cells are usually arranged in nests and separated by thin fibrous and vascular layers and, more rarely, they form trabeculae, islands or at least solid tissue. Features of C cell hyperplasia can be noticed in the surrounding thyroid parenchyma. A characteristic feature, which is not always found, is the presence of amyloid, therefore histopathological diagnosis of medullary carcinoma requires, apart from a classic microscopic test, also an immunohistochemical examination and, first of all, the use of calcitonin antibodies. In over 90% of cases the presence of MTC is connected with a significant increase in calcitonin (Ct) concentration in blood serum. Therefore testing blood for the presence of calcitonin in patients with suspected medullary thyroid carcinoma facilitates its diagnosis.
Medullary thyroid cancer spreads both through the lymphatic system and through the bloodstream. Metastases to lymph nodes are found in 50-75% of cases in the process of diagnosis, often bilaterally with extrafollicular infiltrates. In the process of cancer spreading through the lymphatic system it usually first affects the pretracheal lymph nodes and only then the lateral cervical lymph nodes and it cannot always be seen in an ultrasound scan. The degree of lymph node involvement usually correlates with the size of the primary focus. Metastases to the liver, lungs and bones take place through the blood system.
In the case of regionally advanced medullary thyroid cancer the neoplastic infiltration can spread over the continuity outside the organ's follicle involving vascular-nervous bundles, muscles of the neck as well as the trachea and the oesophagus.
In most patients a thyroid node gradually increasing in size, with varying growth dynamics, usually slow and painless as a rule, is the first symptom of MTC. In several to over 10% of patients diarrhoea occurs which may be the first MTC symptom in some cases and is connected with an advanced form of cancer and it is caused by biologically active peptides and amines being secreted by the tumour. With considerable regional advancement of the diseases, dyspnoea, a sense of obstruction during swallowing or even swallowing disorders occur. Cough, liver enlargement, spontaneous pain and tenderness of palpation in bone, fast weight loss may accompany the disseminated form of the cancer. MTC is a carcinoma where the participation of hereditary predisposal is relatively high and amounts to 20-25% of all cases and in populations in which intense screening tests are performed among family members even over 30% of cases (2, 3).
HEREDITARY MEDULLAR THYROID CARCINOMA
Hereditary MCT may not be accompanied by any other symptoms and in this case we talk about familial medullary thyroid carcinoma (FMTC). However, MCT is more frequently a symptom of multiple endocrine neoplasia type 2 (MEN 2) (tab. 1).
Table 1. Hereditary medullar thyroid carcinoma clinical forms.
| Symptom | FMTC | MEN 2A | MEN 2B |
| medullary thyroid cancer | > 95% | > 95% | > 95% |
| pheochromocytoma | - | ~50% | ~50% |
| parathyroid hyperactivity | - | 15-60% | - |
| typical facial appearance, schwannomas of mucous membrane, hyperplasia of parasympathetic ganglia of the large intestine mucous membrane | - | - | 100% |
Multiple endocrine neoplasia type 2A (MEN 2A), also called Sipple's syndrome, is characterized by a triad of medullary thyroid carcinoma, pheochromocytoma (in approx. 50% of patients), and parathyroid hyperplasia or adenoma (in approx. 15-25% of patients). MCT is usually the first symptom of the syndrome and it becomes manifest during the first two decades of one's life. Pheochromocytomas usually become manifest later and they are rarely the first symptom of the disease. Parathyroid hyperactivity is the last to be revealed, thus the assessment of its occurrence differs depending on the age of patients in the population under analysis.
In atypical forms of MEN 2A it is also accompanied by cutaneous lichen amyloidosis (CLA) or Hirschsprung's disease; however, these syndromes are relatively rare.
Pheochromocytomas are characterized by paroxysmal hypertension accompanied by tachycardia; they may be accompanied by the person paling or excessive perspiration. An undiagnosed/untreated pheochromocytoma can be the cause of sudden death and constitutes even a bigger threat to the patient's life than MCT which may have a non-aggressive course in the MEN 2A type.
Parathyroid hyperactivity leads to elevated calcium levels in blood serum caused by excessive amounts of parathormone. Parathormone enhances bone resorption, hence osteoporosis is an early symptom; symptoms of periosteal resorption or brown tumours occur much later. Features of advanced parathyroid hyperactivity include symptoms of nephrolithiasis, peptic ulcers and pancreatitis may occur. Untreated parathyroid hyperactivity may lead to a hypercalcemic crisis.
As MTC is the first to become manifest, distinguishing between familial MTC and classical MEN 2A requires a longer observation period as pheochromocytomas may become manifest after several years and they will never occur in all family members suffering from MCT. It is generally assumed in the literature that the diagnosis of real familial caner is certain only after at least 4 cases of MCT have occurred in the family and they are not accompanied by a pheochromocytoma or parathyroid hyperactivity. If the number of patients suffering from MTC is lower than 4, we talk about an unclassified form as even DNA test do not enable unambiguous differentiation within this scope (see below).
Diagnosis of MEN 2B is far more unambiguous, both due to a characteristic clinical picture and characteristic mutations. In MEN 2B MTC develops the fastest, already in small children. Pheochromocytomas usually become manifest later and they are revealed in approx. one half of patients, while parathyroid hyperactivity does not occur. Phonotypical characteristics of MEN 2B allow an experienced clinical doctor to diagnose it already during the initial contact with the patient. The patient's appearance is exceptionally characteristic, with an elongated, narrow face, large mandible and very prominent lips. Finding numerous, small schwannomas at the edges of the tongue and the mucous membrane of the oral cavity is a very specific feature during physical examination. In some patients marphanoid features of the body can also be noticed. The hereditary nature of some MTC cases has already been known since the 1960s. Calcitonin tests after pentagastrin administration were used to diagnose cancer among the members of the patient's family (4). Such tests were performed every year among all members of the patient's family until they reached the age of forty. To avoid falsely positive results (pentagastrin can stimulate an increase of calcitonin secretion also in healthy people, especially in young men), an increase in the calcitonin level above 100 pg/ml was treated pathognomonic for hereditary MTC. The determination of calcitonin concentrations enabled a good characterization of genetic predisposal among family members thus facilitating the examination of the link between the occurrence of MTC and genetic markers.
PROTO-ONCOGENE RET AND MEDULLAR THYROID CARCINOMA
In 1987 the gene responsible for hereditary forms of MTC was localized in the centromeric region of chromosome 10. In 1993 it was identified as the proto-oncogene RET and mutations responsible for MEN 2A, FMTC and MEN 2B were described (5, 6, 7).
The proto-oncogene RET encodes receptor tyrosine kinases. In the extracellular part of this membrane protein a region similar to cadherin and a cysteine-rich region located near the cell membrane are found (fig. 1).
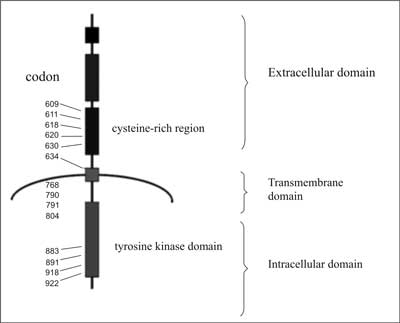
Fig. 1. Diagram of the RET tyrosine kinase receptor structure together with the location of codons undergoing activating mutations.
A short transmembrane part keeps the protein in the cell membrane and in the intracytoplasmic part a domain is located, or actually these are two domains located close to each other, with tyrosine kinase activity. The structure of the protein is closely related to the structure of other receptors for growth factors (e.g. EGF) which are actually receptor tyrosine kinases. A small neuropeptide – glial cell-derived neurotrophic factor (GDNF) is the ligand responsible for the transdution of the growth signal. This peptide is not directly connected with the RET protein but with another membrane protein, called the α-receptor for GDNF (at present GFR α-1), fulfilling the function of an RET co-receptor (fig. 2). Autophosphorylation of the receptor is a consequence of its activation which induces a cascade of MAP kinases and transcription of genes participating in cell proliferation.
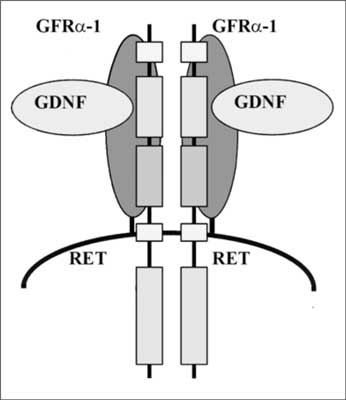
Fig. 2. Physiological activation of tyrosine kinase RET.
Mutations of the RET proto-oncogene, leading to the development of MCT have the nature of mutations activating the protein product function (2). The RET proto-oncogene consists of 21 exons. However, mutations occur in only a few of them and they are usually point mutations (their localization relative to the encoded protein is shown in figure 1). They usually concern the codons encoding cysteines in the extramembrane part of the receptor, close to the cell membrane. Codon 634, located in exon 11 and constituting part of the transmembrane section undergoes mutation most frequently (in 75-80% of all cases of hereditary MTC) (tab. 2). Most mutations within this codon (over 90%) are mutations resulting in changing cysteine to arginine, tyrosine or tryptophan (8, 9).
Table 2. Localization of proto-oncogene RET mutations causing hereditary medullary thyroid carcinoma (10, 11).
| Codon/Exon | Type | Frequency of occurrence (13) | Frequency of occurrence (%) (39) |
| 609/10 | MEN 2A/FMTC
MEN 2A/ch.
Hirschsprung's | 0-1 | 0% |
| 611/10 | MEN 2A/FMTC | 2-3 | 2,5% |
| 618/10 | FMTC/MEN 2A
MEN 2A/ch.
Hirschsprung's | 3-5 | 12% |
| 620/10 | FMTC/MEN 2A
MEN 2A/ch.
Hirschsprung's | 6-8 | 3% |
| 630/11 | MEN 2A/FMTC | 0-1 | 0% |
| 634/11 | MEN 2A
MEN 2A/CLA | 75-85 | 42% |
| 635/11 | MEN 2A | rarely | not tested |
| 637/11 | MEN 2A | rarely | not tested |
| 768/13 | FMTC | 0-1 | 1% |
| 790/13 | FMTC/MEN 2A | 0-1 | 2,5% |
| 791/13 | FMTC | 0-1 | 16% |
| 804/13 | MEN 2A/FMTC | 0-1 | 8% |
| 883/15 | MEN 2B | rarely | rarely |
| 891/15 | FMTC | rarely | not tested |
| 918/16 | MEN 2B | 3-5 | 12% |
| 922/16 | MEN 2B | rarely | not tested |
Classic MEN 2A is most likely if a mutation within codon 634 occurs, while other mutations are connected with a significantly lower probability of the development of a pheochromocytoma – as a result of the mutation the familial MTC usually develops without other endocrinopathies (tab. 2).
Mutations within codon 918 (exon 16) apply to the tyrosine kinase domain. As different cell proteins undergo phosphorylization, the phenotype of MEN 2B is different from the phenotype of MEN 2A. Excessive activation of RET can also be observed in peripheral nerves (schwannomas of the tongue and the mucous membrane of the oral cavity and the lips, hypergangliosis of the large intestine), medullary cancer becomes manifest earlier and its course is more aggressive, however, no parathyroid hyperplasia occurs (12, 13).
Mutations in codons 768, 790, 791, 804 and 891 also concern the intracellular part of the RET protein (14, 15, 16, 17), they occur rarely and their transformational potential is rather small – apart from the mutation in codon 790 which was demonstrated in both FMTC and MEN 2A (14), they mostly lead to the development of familial RRT which may become manifest relatively late – often only in the fourth or fifth decade of life. Regarding mutations in codon 791 it is supposed that its penetration may not be full. Other mutations of the RET gene are characterized by nearly full penetration – thus the finding of a germinal mutation is tantamount to an over 90% certainty of developing RRT. In families with the RET 791 mutation a considerable changeability of risk occurs and at least in some of them full-blown RRT becomes manifest relatively early.
THE GENOTYPE-PHENOTYPE DEPENDENCY IN THE HEREDITARY FORM OF MEDULLARY THYROID CARCINOMA
In hereditary MTC the dependency between the localization of the RET point mutation and the clinical picture of the disease can be clearly distinguished.
From the genetic point of view, MEN 2A and familial MCT are close to each other and at present familial MCT is treated as one of the forms of MEN 2A. MEN 2B is regarded as separate as it has its own typical mutations and its own typical phenotype. The probability of the occurrence of typical MEN 2A heavily depends on the localization of the mutation – it is very high, if the mutation occurs within codon 634 (the full syndrome with parathyroid hyperactivity occurs especially often when cysteine is replaced with arginine), it is lower in exon 10 and it is very low, if the mutation occurs in exons 13 and 15 (tab. 2) (18).
DNA TESTS
As hereditary cancer constitutes a significant part of all cases of MCT and may not be clinically different from non-hereditary cancer, full molecular tests for the presence of germinal mutations must be conducted in all patients with such a diagnosis. Even if not other features of MEN 2 occur in a patient and the medical history in their family is negative, the risk of detecting a germinal mutation is approx. 10% in our population (3). In figure 3 an algorithm of searching for mutations in the proto-oncogene RET (19, 30). The sequence of testing the codons of the RET proto-oncogene depends on the frequency of the mutation occurrence. Hence, it starts from testing codon 634. This test may be performed by means of the PCR/RFLP technique. Nevertheless, a negative result of this test does not mean that it is not necessary to perform tests for other known mutations, as nearly a half of newly diagnosed cases of hereditary cancer concern mutations in exons 13-15. For this reason DNA tests should also be performed after diagnosing MCT in older people – as it has already been mentioned, some mutations are characterized by a distinctly later manifestation of MCT.
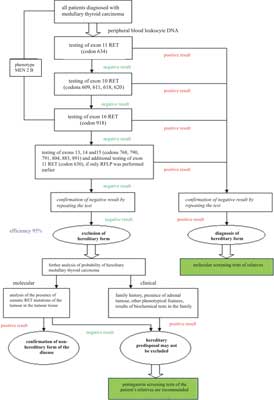
Fig. 3. Algorithm of DNA diagnostics in patients suffering from medullary thyroid carcinoma.
Tests of mutations in exons 10 and 13-15 require sequencing. Tests for mutations in codon 918 are generally performed on the basis of information about the phenotype, although in some cases phonotypical features of MEN 2B may be weakly marked and only molecular tests make it possible to make a correct diagnosis. It needs to be emphasized that the small size of the gene and a limited number of characteristic mutations are factors facilitating DNA testing. Such tests are performed at several centres in our country*. The identification of a germinal mutation in an MCT patient means significant advantages for the patient and their family. In the patient it makes it possible to determine the risk of pheochromocytoma and parathyroid hyperactivity occurrence, thus determining the frequency of screening tests. At the same time it shows hereditary predisposal in the patient and is an absolute indication for the implementation of DNA testing for the members of the patient's family. The risk of detecting mutations among first-degree relatives is 50%. Our research shows that on average one mutation carrier is identified for each newly diagnosed case of hereditary MCT (3, 21). The probability of early detection of cancer in a mutation carrier changes depending on the mutation type and the age of the family members – in some of them we already deal with clinically detectable thyroid cancer, while in others we observe an increase in calcitonin concentrations either in the initial test or after stimulation with pentagastrin but without a node noticeable in an ultrasound scan of the thyroid. If tests are implemented early it is possible to identify a carrier at a symptom-free stage.
The negative result of the test for germinal mutations is equally important. It makes it possible to exclude a given family member from further check-ups, if no mutation characteristic of a given family is found. The negative result of DNA tests performed after diagnosing medullary cancer in order to detect an hereditary form has a predictive value of approx. 90% (2), as there exist families (it particularly applies to familial MTC) in which no germinal mutation was found despite several cases of medullary cancer. Thus, if the medical history of a family or an individual is positive and the DNA test result is negative, the continuation of annual pentagastrin tests in the entire family is the only solution.
BIOCHEMICAL TESTS APPLIED IN DIAGNOSING AND MONITORING MEDULLARY THYROID CARCINOMA AND MEN 2
MCT cells usually secrete large amounts of calcitonin. Its determination allows preoperative detection of cancer and it is also a good tool for the assessment of the effectiveness of the treatment applied and the monitoring of the further course of the disease (22, 23, 24). At present, routine performance of pentagastrin tests in members of the patient's family is not needed as the hereditary form of the disease can be identified by means of DNA tests. The determination of calcitonin levels should be performed at a specialized laboratory, the standard range of good tests is up to 10 or more than 10 pg/ml. The determination of calcitonin concentration is a necessary element of the effectiveness of the applied treatment of medullary thyroid carcinoma. The normalization of the elevated preoperative hormone concentration to normal values after the operation confirms its radicality. The persistence of incorrect values, despite the macroscopic and microscopic radicality of the procedure, implies that microfoci of cancer are present in the lymph nodes.
Low concentrations of the hormone (below 10-12 pg/ml) in check-up tests performed every 3 months imply complete regression of the tumour. Nevertheless, it is still necessary to perform a provocative test once a year. Intravenous infusion of calcium, pentagastrin or oral administration of omeprasole is used for this purpose. The pentagastrin test is applied most frequently. The determination of calcitonin concentration is conducted using samples collected 2, 5 and 10 minutes after pentagastrin administration (0.5 ?g per kilogram of body weight). An increase in the calcitonin concentration exceeding 30 pg/ml confirms the presence of cancerous cells.
A high calcitonin concentration is an indication for imaging tests in order to localize the focus of the cancer. If their result is negative, catherization of cervical and hepatic veins may be considered. An increase in calcitonin concentration in the specified sample after pentagastrin administration makes it possible to locate a recurrence or a metastasis within the vessels supplying the vein from which the sample was collected.
Normal concentrations of calcitonin are relatively rarely observed in patients with clinical symptoms of medullary cancer. It may result from a low level of tumour differentiation and the loss of hormonal activity in some patients. It also needs to be remembered that high antigen concentrations (i.e. calcitonin in this case) can inhibit its binding to the antibody used in the radioimmunological test, and, in consequence, a falsely negative result is obtained (the so-called "hook effect”). To detect this phenomenon it is enough to test the serum after dilution. Serum dilution is necessary in many cases anyway, as calcitonin concentrations observed in patients suffering from medullary thyroid carcinoma may exceed the range of determinable concentration in available immunometric tests by 10-1000 times.
Other cancer markers are less significant for medullary cancer. The CEA test is used most frequently. An elevated CEA level implies considerable advancement of the disease.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Hazard JB, Hawk WA, Crile G Jr: Medullary (solid) carcinoma of the thyroid – a clinicopathology entity. J Clin Endocrinol Metab 1959; 19: 152-61.
2. Eng C: RET proto-oncogene in the development of human cancer. J Clin Oncol 1999; 17 (1): 380-93.
3. Wiench M, Wygoda Z, Gubała E et al.: Genetic diagnosis of multiple endocrine neoplasia type 2B. Endokrynologia Polska 2000; 51: 67-76.
4. Barbot N, Calmettes C, Schuffenecker I et al.: Pentagastrin stimulation test and early diagnosis of medullary thyroid carcinoma using immunoradiometric assay of calcitonin: comparison with genetic screening in hereditary medullary thyroid carcinoma. J Clin Endocrinol Metab 1994; 78: 114-20.
5. Donis-Keller H, Dou S, Chi D et al.: Mutations of the RET proto-oncogene are associated with MEN 2A and FMTC. Hum Mol Genet 1993; 2: 851-6.
6. Hofstra RM, Landsvater RM, Ceccherini I et al.: A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 1994; 367: 375-6.
7. Mulligan LM, Kwok JB, Healey CS et al.: Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 1993; 363: 458-60.
8. Eng C, Clayton D, Schuffenecker I et al.: The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET Mutation Consortium analysis. JAMA 1996; 276: 1575-9.
9. Eng C, Mulligan LM: Mutations of the RET proto-oncogene in the multiple endocrine neoplasia type 2 syndromes, related sporadic tumors, and Hirschsprung disease. Human Mutation 1997; 9: 97-109.
10. Gagel RF, Cote GJ: Pathogenesis of medullary thyroid carcinoma. Thyroid Cancer. Kluwer Academic Publisher. Boston/Dordrecht/ London 1998.
11. Brandi ML, Gagel RF, Angeli A et al.: Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001; 86: 5658-71.
12. Gimm O, Sutter T, Dralle H: Diagnosis and therapy of sporadic and familial medullary thyroid carcinoma. J Cancer Res Clin Oncol 2001; 127: 156-65.
13. Kitamura Y, Goodfellow PJ, Shimizu K et al.: Novel germline RET protooncogene mutations associated with medullary thyroid carcinoma (MTC): mutation analysis in Japanese patients with MTC. Oncogene 1997; 14: 3103-6.
14. Berndt I, Reuter M, Saller B et al.: A new hot spot for mutations in the RET protooncogene causing familial medullary thyroid carcinoma and multiple endocrine neoplasia type 2A. J Clin Endocrinol Metab 1998; 83: 770-4.
15. Bolino A, Schuffenecker I, Luo Y et al.: RET mutations in exons 13 and 14 of FMTC patients. Oncogene 1995; 10: 2415-9.
16. Eng C, Smith DP, Mulligan LM et al.: AJ novel point mutation in the tyrosine kinase domain of the RET proto-oncogene in sporadic medullary thyroid carcinoma and in family with FMTC. Oncogene 1995; 10: 509-13.
17. Hofstra RM, Fattoruso O, Quadro L et al.: A novel point mutation in the intracellular domain of the RET protooncogene in a family with medullary thyroid carcinoma. J Clin Endocrinol Metab 1997; 82: 4176-8.
18. Wohllk N, Cote GJ, Bugalho MM et al.: Relevance of RET proto-oncogene mutations in sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 1996; 81: 3740-5.
19. Jarząb B, Włoch J, Wirnch M et al.: Wczesna diagnostyka zespołu mnogiej gruczolakowatości wewnątrzwydzielniczej typu 2 poprzez analizę genetyczną germinalnych mutacji protoonkogenu RET. Endokrynol Pol 1999; 50: 127-34.
20. Lips CJ, Landsvater RM, Höppener JW et al.: Clinical screening as compared with DNA analysis in families with multiple endocrine neoplasia type 2A. N Eng J Med 1994; 331: 828-35.
21. Włoch J, Wygoda Z, Wiench M et al.: Profilaktyczne całkowite wycięcie tarczycy u nosicieli mutacji w protoonkogenie RET powodujących dziedzicznego raka rdzeniastego tarczycy. Pol Przegl Chir 2001; 73: 569-85.
22. Krassowski J et al.: Oznaczanie kalcytoniny w rozpoznawaniu i ocenie wyników leczenia raka rdzeniastego tarczycy. Pol Tyg Lek 1989; 44: 757-9.
23. Pacini F, Fontanelli M, Fugazzola L et al.: Routine measurement of serum calcitonin in nodular thyroid diseases allows the preoperative diagnosis of unsuspected sporadic medullary thyroid carcinoma. J Clin Endocrinol Metab 1994; 78: 826-9.
24. Wasylewski A et al.: Przydatność oznaczania kalcytoniny w ocenie doszczętności zabiegu operacyjnego u chorych z rakiem rdzeniastym tarczycy. Endokrynol Pol 1981; 32: 239-44.
25. Januszewicz A: Nadciśnienie tętnicze. Zarys patogenezy, diagnostyki i leczenia. Medycyna Praktyczna 2002.
26. Rekomendacje Diagnostyka i Leczenie raka tarczycy przyjęte podczas III Konferencji naukowej. Rak tarczycy., Szczyrk, 25.03.2006 Endokrynol Pol 2006; 57: 458-77.
27. Januszewicz A, Januszewicz W, Jarząb B et al.: Wytyczne dotyczące diagnostyki i leczenia chorych z guzem chromochłonnym. Nadciśnienie tętnicze 2006; 10: 1-19.
28. Włoch J: Postępowanie chirurgiczne w dziedzicznym raku rdzeniastym tarczycy: modyfikacje wynikające z diagnostyki mutacji germinalnych protoonkogenu RET i badania profilu molekularnego guzów (rozprawa habilitacyjna). Nowotwory (w druku).
29. Dralle H, Gimm O, Simon D et al.: Prophylactic thyreoidectomy in 75 children an adolescent with hereditary medullary thyroid carcinoma: German and Austrian experience. World J Surg 1998; 22: 744-750.
30. Skinner M et al.: Prophylactic thyroidectomy in multiple endocrine neoplasia type 2A. N Engl J Med 2005; 15, 353: 1105-13.
31. Baudin E, Travagli JP, Schlumberger M: How effective is prophylactic thyroidectomy in asymptomatic multiple endocrine neoplasia type 2A? Nat Clin Pract Endocrinol Metab 2006; 2: 256-7.
