© Borgis - Postępy Nauk Medycznych 11/2010, s. 860-865
*Stanisław Zajączek
Neurofibromatosis typ 1 (NF1, von Recklinghausen disease)
Nerwiakowłókniakowatość typu 1 (NF1, choroba von Recklinghausena)
International Hereditary Cancer Center, Genetics and Pathology Unit, Cytogenetics Unit, Department of Pathology, Pomeranian Medical University, Szczecin
Head of the Genetics and Pathology Unit: prof. dr hab. med. Jan Lubiński
Head of the Cytogenetics Unit: prof. dr hab. Stanisław Zajączek
Streszczenie
Neurofibromatoza 1 jest częstą chorobą autosomalną dominującą (1:3500 ż.u.), ekspresja jest indywidualnie zmienna i zależna od wieku. Około połowa przypadków jest sporadyczna i wynika z mutacji de novo; jednak ryzyko u ich potomstwa wynosi 50%. Znaczna wielkość genu neurofibrominy uniemożliwia rutynową diagnostykę molekularną, która jest zarezerwowana dla przypadków diagnostycznie trudnych (n. segmentowa, mozaiki, fenotypy z niepełną penetracją lub ekspresją). Rozpoznanie ustala się w oparciu o kryteria kliniczne ujęte w NF1 „NIH Consensus Conference Criteria”. Ekspresja objawów jest indywidualnie i rodzinnie zmienna.
Przedstawiono zasady diagnostyki, opieki i oceny ryzyka nowotworowego. Poza zmianami skórnymi rozwijają się także glejaki okolicy n. wzrokowego (astrocytoma pilocyticum) i rozlane hiperintensywne ogniska T2 w MRI OUN. Towarzyszyć im może padaczka, zaburzenia hormonalne i widzenia, deficyty psychospołeczne. Częste są dysplazje i wady szkieletu.
Mutacje NF1 zwiększają ryzko nowotworów, jednak nie są znane dokładne wartości ryzyka. Wysokie ryzyko dotyczy guzów wywodzących się z otoczki nerwów obwodowych (MPST) i białaczek. Te ostatnie opisywane są jako postaci białaczki nie – limfocytowej o wczesnym początku; przebiega ona ze złym rokowaniem.
Przedstawiono rzadkie zespoły chorobowe skojarzone z NF1 i ich różnicowanie. Mutacje opisanego niedawno genu SPRED są odpowiedzialne za zespół łączący objawy zespołu Noonan i NF1, klasyfikowane poprzednio jako forma kliniczna typowej NF1.
Summary
Neurofibromatosis 1 is a relatively frequent autosomal dominant disease (1:3500 live births), its expression depends on the and on an individual. 50% of the cases results from de novo mutations and are non-familial, but NF1 manifests in following generations with a 50% risk. Due to the great size of the neurofibromin gene, molecular analysis is not routinely employed and is reserved primarily for cases difficult to diagnose (e.g. segmental form, mosaics, not fully penetrating and expressing phenotypes). The disease is diagnosed based on the analysis of defining clinical features, summarized in 1997 in NF1 NIH Consensus Conference Criteria. expression of the symptoms individually and familial varies. The following section presents the principles of diagnosis, familial care and neoplasia risk determination. In addition to distinguishable skin lesions, patients develop gliomas near the optic pathways, and diffuse hyperintensive foci of the MRI T2 signal in CNS. Gliomas are morphologically presented as astrocytoma pilocyticum. NF1 may be associated with epilepsy, visual and hormonal disorders and psychosocial deficits. Skeletal dysplasias and deformations occur frequently. NF1 mutations increase the risk of neoplasia however, risk profiles remain unknown. High risk is linked with MPNST – Malignant Peripheral Nerve Sheath Tumors and leukemia, described as Juvenile Onset Non-Lymphocytic Leukemia (refractory and with a poor prognosis). Additionally presented are rare similar syndromes difficult to diagnose. Mutations of the newly discovered gene SPRED are responsible for a different syndrome, combining signs of Noonan Syndrome and NF1, previously classified as the clinical form of NF1.

Overview
Neurofibromatosis 1 is a relatively frequent autosomal dominant disease with an incidence of about 1 in 3000 births. It is considered one of the phakomatoses; (from Greek phakos "marked at birth”). This group comprises various diseases, involving different primary germ cell layers and manifesting complex signs on the skin, CNS and vision disorders. Lesions detected in a course of NF1 are mainly of neuroectodermal origin (6, 8, 11, 14).
NF1 remains undiagnosed in a significant number of cases. The culprit behind this is the typical textbook image of patients with the final stage of the skin signs, while NF1 in most cases has a mild course and symptoms can vary from person to person in affected families, in people carrying identical mutations and in an individual over the course of time (11).
Signs and diagnosis
NF1 diagnosis is mainly based on physical examination with an evaluation of major and minor diagnostic criteria, listed below. In evident cases molecular diagnostics is not necessary to establish the diagnosis of NF1.
Both major and minor diagnostic criteria combine in a range of constellations and with diverse severity even among patients from the same family.
Clinical manifestations tend to evolve with age, particularly in children. The diagnosis can typically be given at the age of 8 to 14 years. Probable lack of direct genotype-phenotype correlation makes individual prognosis prediction virtually impossible.
Current diagnostic criteria were developed in 1997 under NF1 NIH Consensus Conference Criteria (8). According to it, a positive diagnosis of NF1 requires at least two of the following features to be present:
? 6 or more café-au-lait spots, with over 5 mm in the greatest diameter, in pre-pubertal individuals and with over 15 mm in the greatest diameter in post-pubertal individuals.
? Two or more neurofibromas of any type or one plexiform neurofibroma.
? Freckling in the axillary or inguinal regions.
? Optic glioma.
? Two or more Lisch nodules.
? Distinctive osseous lesions.
? A first-degree relative with type 1 neurofibromatosis according to the above criteria. [Huson and Korf (2)].
Single café-au-lait spots are present in approximately 10-15% of healthy children and are detectable after childbirth; lack of café-au-lait spots after puberty precludes NF1 diagnosis (8, 11, 14).
Practically all patients with NF1 have cutaneous neurofibromas, localized in both the dermis and epidermis, manifesting as "rubbery” and are passively movable to some extent. They are normally not painful, and are occasionally itchy. Neurofibromas may, with time, evolve into plexiform subtype and trigger dramatic deformations as in classical NF1 descriptions. Plexiform neurofibromas, in 5% of patients, localize near nerves giving neurological deficits and causing pain (8, 11, 14).
Lisch nodules are hamartomas of the iris, they are not sight threatening but have a valuable diagnostic feature. The lesions should be evaluated using slit lamps (11, 14). Plexiform neurofibromas may infiltrate numerous nerve sheaths and branches. Large subdermal nodules that reach a few centimeters in size grow faster in the first years of life, while later tend to stabilize. Skin above the lesions is typically affected, nodules may cause pain, itching sensation and other symptoms resulting from nerve and bone pressure. Although nodules may undergo a malignant transformation, the majority of sarcomas develop in regions without typical NF1 lesions (11).
Optic and brain gliomas, as well as diffusion of hyperintensive foci of MRI T2 signal, occur in childhood in 15% of patients. Gliomas are morphologically presented as astrocytoma pilocyticum and develop along the optic pathways leading to signs of proptosis, lowered visual acuity and loss of visual field. Relatively frequent is hypothalamic localization triggering endocrinopathies, e.g. GH deficiency (medical treatment with rh-GH in patients with hypothalamic disorders and NF1 is nonetheless disputable) and precocious puberty. Refractory epilepsy is a manifestation of gliomas in certain brain areas. Therapeutic decisions concerning glioma are particularly complicated because spontaneous regression or stabilization is occasionally observed, yet it is unpredictable (11).
A significant number of patients with NF1 present bone dysplasia and malformation, primarily thoracic scoliosis (5). In our observation the above mentioned disorders are commonly associated with osteopenia.
Approximately 1/3 of patients show psychosocial deficits; severe and moderate mental retardation occurs in below 1% of patients. In 30 to 60% of patients subtle visual-spatial disorientation, dyslexia, short-term memory dysfunction and inferior verbal test results in IQ test are found (1, 5, 6, 8, 11, 13).
Molecular basis of NF1
NF1 is an autosomal dominant disease with penetrance of up to 100% and highly variable mutation expression of the NF1 gene (neurofibromin protein) (2, 4, 11, 17, 20).
50% of NF1 cases arise due to spontaneous mutation without familial background; children of those patients inherit NF1 with typical 50% risk (1, 3, 4).
Neurofibromatosis I is a result of germline or (rarely) somatic mosaics, an inactivating mutation of the neurofibromin gene (OMIM 162200) located at chromosome 17p11.2 and resulting in the haploinsufficiency of their product.
NF1 gene is a tumor suppressor. Coding protein – neurofibromin – is a negative regulator of the RAS oncogene and its cascade. It stimulates active guanosine 5" – triphosphate RAS conversion to inactive diphosphate form, and indirectly regulates the activity of certain growth factors. Other related diseases, e.g. Noonan, Costello and Leopard syndromes result from RAS regulators cascading mutations and form a distinguishable syndrome complex.
NF1 gene spans 280 kb, codes ~9 open reading frames and comprises 61 exons, 4 of which are alternatively spliced. It includes two large introns (1 and 27b), the latter containing three small unrelated genes EV12A, EV12B and OMG, each encompassing two exons and transcribed in reverse orientation. Its possible role and interference with NF1function is unknown. In various tissues NF1gene may be alternatively transcribed. Exons 9a, 10a-2, 23a and 48a are spliced alternatively but this processes does not disrupt the overall reading frame. There is a great number of pseudogenes, localized in various human chromosomes. See figure 1 (4, 20).
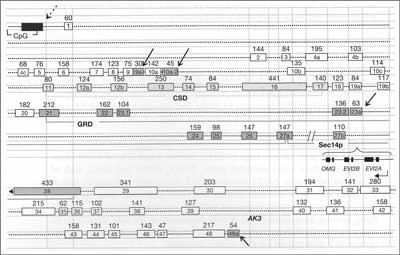
Fig. 1. Known Structure of the NF1 gene. Exons are shown as a numbered rectangles with size indicated (Bp). Four alternatively spliced exons are shown as a blue boxes (arrows). Introns are draft as a dotted line. Promoter is shown as a blue box (dotted arrow) with CpG islands. Main recognized neurofibromin domains are noted as CSD (Cysteine – Serine Rich), GRD (GTPase Activating Related), Sec 14p (homologous to Saccharomyces protein). Three embeeded genes in intron 27 (OMG, EV12A, EV12B) are transcribed in opposite direction. AK3 is a Adenylate Kinase pseudogene. Acc. to Upadhyaya 2008 (20).
All known mutations type were detected in the NF1 gene; the vast majority are stop mutations leading to lack of the protein or its non-functional truncations. Due to the great size of the gene PTT, test and certain variants of RNA assays may be more practical. Mutations are localized mostly without any preferential "hot-spots”, except for sequences near highly methylated and highly repetitive regions. Typically mutations have a recurrence rate of approximately 0.5-1.0% in patient mixed population.
The NF1gene is flanked by two sets of paralogs NF1- REP-P and NF1REP-M, flanked by 46 bp JJAZ1gene and its pseudogene – Ν. Homologous recombination between these sites generate two types of recurrent 1.2 and 1.4 Mb microdeletion, containing in both cases all of the NF1 gene. 1.4 Mb deletion, detected in ~5% of NF1 patients, comprising not only NF1, but also 14 other genes. Their phenotype corresponds to contiguous gene syndrome definition and creates a special subgroup of NF1 patients. Its phenotypes are characterized by severe mental disability, facial dysmorphies, cardiac pathologies, growth disturbances, an earlier onset with a large number of cutaneous neurofibromas and a greater, compared to typical patients, risk of malignancy. Microdeletions may be diagnosed quickly and simply by FISH (fig. 2, 3) (4, 17).
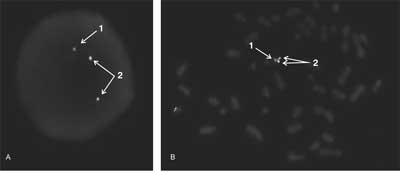
Fig. 2. Konstitutional large deletion of the NF1 gene detected by FISH (Cytocell Probe LPU017). Red signal (1) – only one copy of the NF1 gene, green (2) – two control signals from the other part of chromosome 17. Only one copy of the genes NF1 are detected in interphase (A) and metaphase (B) of peripheral lymphocytes (phot. Ewa Studniak, Cytogenetics Unit, PUM).
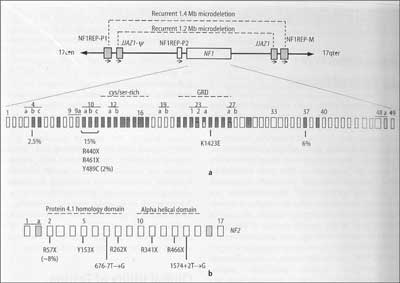
Fig. 3. Schematic presentation of NF1(a) and NF2(b) genes, to explain thr mechanisms of generation of the large deletions of NF1 gene. Acc. to Stephens, 2008 (18).
Malignancies and NF1
Patients with NF1 have an increased risk of developing malignant neoplasms including mainly rhabdomyosarcomas, MPNST (Malignant Peripheral Nerve Sheath Tumors) and leukemia. The majority of CNS lesions are astrocytomas.
Differentiation between benign T2 lesions and gliomas in MRI requires repeated examinations performed by experienced neuroradiologist. MPST are highly aggressive sarcomas developing near plexiform neurofibromas (6, 8, 11).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Benjamin CM, Colley A, Donnai D et al.: NF1: Knowledge, Experience and Reproductive Decisions of affected Patients and Families. J Med Genet 1993; 30: 567-94.
2. Bernards A: Neurofibromatosis type 1 and Ras – Mediated Signalling: Fitting in the GAP"s, Bioch Bioph Acta 1995; 1242: 43-59.
3. Brems H, Chmara M, Sabbatou M et al.: Germline Loss of Function Mutations In SPRED1 Cause Neurofiromatosis 1 Like Phenotype. Nat Genet 2007; 39: 1120-6.
4. Evans DGE, Wallace A: "NF2: Mutations and Menagement of Disease”. In Neurofibromatoses, Kofmann D. (ed.) Monogr. In Hum. Genet 2008; 16, 154-166: Basel, Karger
5. Friedmann JM, Birch P, Greene C: NNFF International Database Participants: National Neurofibromatosis Foundation International Database. Am J Med Genet 1993; 45: 88-91.
6. Friedmann JM: Neurofibromatosis type 1 Gene Clinics, http://www.geneclinics. org/profiles/nf1/details.html (2010).
7. Goldberg MJ: The Dysmorphic Children – An Orthopedic Perspective: VI. Neurofibromatosis and the Historical Phakomatoses, Raven Press NY 1987; 225-46.
8. Gutmann DH, Consensus Group: The diagnostic Evaluation and Multidisciplinary Menagement of Neurofibromatosis 1 and Neurofibromatosis 2, J Am Med Ass 1997; 278: 51-7.
9. Gusella J: Neurofibromatosis at the Millenium, The National Neurofibromatosis Foundation Millenium Lecture 2000; http://www.nf.org
10. Griffiths DF, Williams GT, Williams ED: Multiple Endocrine Neoplasia Associated with von Recklinghausen Disease. Br Med J 1983; 287: 1341-3.
11. Huson SM, Korf B: Phakomatoses [In:] Emery"s and Rimoin"s Principles and Practice of Medical Genetics, Churchill-Livingstone, London 2002; 3: 3162-202.
12. Maris JM, Wiersma SR, Matgoub N et al.: Monosomy 7 Myelodysplastic Syndrome and Other Second Malignant Neoplasms in Children with NF type 1. Cancer 1997; 79: 1438-46.
13. North K, Joy MA, Yuille D et al.: Specific Learning Disability in Children with Neurofibromatosis type 1: Significance of MRI Abnormalities, Neurology 1994; 44: 878-83.
14. Riccardi VM: Von Recklinghausen Neurofibromatosis. N Engl J Med 1981; 305: 1617-27.
15. Riccardi VM: Neurofibromatosis: Phenotype, Natural History and Pathogenesis, 2nd Ed J Hopkins Univ Press, Baltimore 1986.
16. Ricciardone M, Ozcelik T, Cevber B et al.: Human MLH1deficiency Predisposes to Hematological Malignancy and NF Type I. Cancer Res 1999; 59: 290-3.
17. Shannon KM, Watterson J, Johnson P et al.: Monosomy 7 Myeloproliferative Disease in Children with NF Type 1: Epidemiology and Molecular Analyses. Blond 1992; 79: 1311-8.
18. Stephens KL: The Neurofibromatoses. [In:] Leonard D, Kaul K, Van Deerlin V. (eds.) Molecular Pathology in Clinical Practice – Genetics. Springer Verl., NY 2008; pp. 239 +256.
19. Tartaglia M: Gain of Function SOS1 Mutations Cause a Distictive Form of Noonan Syndrome. Nat Genet 2007; 39: 75-9.
20. Upadhyaya M: NF1 Gene Structure and NF1 Genotype/Phenotype Correlations. [In:] Neurofibromatoses, Kofmann D. (ed.) Monogr. In Hum. Genet 2008; 16, 46-62: Basel, Karger.
21. Wang Q, Lasset C, Desseigne F et al.: Neurofibromatosis and Early Onset of Cancers in hMLH1-Deficient Childrens. Cancer Res 1999; 59: 294-7.
22. Zajączek S, Peregud-Pogorzelski J, Cybulski C et al.: Juvenile Onset Chronic Myeloid Laeukemia in a Girl with the Familial Form of NF-1. Clin Genet 1999; 11: 477.
23. Zvulunov A, Barak Y, Metzger A: Juvenile Xanthogranuloma, Neurofibromatosis and Juvenile Chronic Myelogenous Leukemia. Arch Dermatol 1995; 131: 904-8.
