© Borgis - Postępy Nauk Medycznych 11/2010, s. 854-859
*Stanisław Zajączek
Retinoblastoma
Siatkówczak
International Hereditary Cancer Center, Genetics and Pathology Unit, Cytogenetics Unit, Department of Pathology, Pomeranian Medical University, Szczecin
Head of the Genetics and Pathology Unit: prof. dr hab. med. Jan Lubiński
Head of the Cytogenetics Unit: prof. dr hab. Stanisław Zajączek
Streszczenie
Siatkówczak jest modelowym nowotworem o podłożu genetycznym. Warunkowany jest dominującymi mutacjami jednej kopii genu RB1, które mogą mieć charakter somatyczny (S. sporadyczny, zwykle jednostronny) lub konstytucyjny (często obustronny i/lub wieloogniskowy). Do powstania guza konieczne jest jeszcze „drugie trafienie”, które ma już charakter mutacji somatycznej. Unieczynnienie obu kopii genu odblokowuje cykl komorkowy w fazie G1-S i inicjuje nowotwór. Gen RB1 składa się z 27 eksonów, mutacje nie wykazują preferencji lokalizacyjnych typu hot spot (co utrudnia diagnostykę), jednak ostatnio opisano pewne preferencje lokalizacyjne i etniczne w charakterystyce mutacji. Omówiono uwarunkowania genetyczne i charakterystykę rodowodowo-kliniczną siatkówczaka. Podano zasady diagnostyki genetycznej siatkówczaka a także zasady opieki nad rodzinami obciążonymi zmutowanym genem. Z uwagi na występowanie nosicieli mutacji konstytucyjnych również w grupie pacjentów z guzami jednostronnymi sporadycznymi, wszyscy chorzy z siatkówczakiem jak i ich rodzeństwo powinni być poddawani badaniom profilaktycznym do chwili potwierdzenia/wykluczenia mutacji. Nosicielstwo mutacji konstytucyjnej powoduje także w późniejszym życiu pacjenta podwyższone ryzyko innych pierwotnych nowotworów, zwłaszcza mięsaków.
Summary
Retinoblastoma genetics is a model system for gaining knowledge on the basic features of all hereditary cancers. A tumor is caused by inactivating mutations in both copies of the RB1 gene. They are transmitted as "one hit” in a dominant manner, but an inactivation of the second gene copy is needed for initiation. In hereditary form (frequently bilateral/multifocal) the first "hit” is constitutional and inherited but a second hit is a somatic mutation. In sporadic form (mostly unilateral and unifocal) both "hits” are somatic mutations. Inactivation of both RB1 gene copies abolishes the cell cycle block between G1 and S phase initializing carcinogenesis. The RB1 gene consists of 27 exons, mutations do not exhibit hot spots, but some spatial and ethnic regularities were detected in recent years. Hereditary determination, clinical value of pedigree and molecular analysis of the RB1 gene are described, as well as basic rules of familial care. Due to the detection of constitutional mutations in some sporadic/unilateral cases, carrier status in some patients with non-familial, unilateral tumors ( de novo and germ-line or low penetrant mutations) prophylactic investigations of all first degree relatives must be performed, until their molecular verification is done. Since a patient with a sporadic, unilateral tumor might be a constitutive mutation carrier, all patients with retinoblastoma and their siblings should be tested until the mutation is confirmed or excluded.
Patients with constitutional mutations also have a higher risk of secondary primary tumors, particularly sarcomas.

Retinoblastoma (Rb) plays a significant role among tumors. It is the first neoplasm with the hereditary etiology that has been demonstrated. It helped in developing the "two-hits” hypothesis and the suppressor gene idea (4, 5, 15, 17).
The incidence of Rb is 1 in 25,000 of live-born infants. Despite its rarity, Rb is the most common intraocular neoplasm in children. In the majority of cases it is diagnosed before the age of 5 years. Rb scarcely occurs in adults. 60% of cases of Rb show a true sporadic character. These are the results of somatic mutations occurring in the retinal cells. The remaining 40% of patients are children with constitutive mutations. Among them 10-15% are familial cases and the remaining 25-30% are new in the family as the result of a de novo germinal constitutive mutation. The penetrance of the constitutive mutation reaches 90%. For the onset of neoplasm the inactivation of protein pRB is needed. It occurs after the function of both alleles has been lost or inactivated (17).
The probability of the appearance of two consecutive somatic mutations in the RB1gene in the same retina cell, as it happens in the sporadic form of Rb, is directly proportional to the duration of cell life. It explains the late onset and usually unilateral and unifocal development of such forms of Rb in comparison with Rb caused by constitutive mutation (fig. 1). According to the "two-hits” hypothesis the development of the tumors in patients with a constitutive mutation requires only a next simple somatic mutation in retina cell. This is more likely to happen earlier, compare to two accidental sporadic mutation, thus the average age of patients with hereditary form of Rb is lower. Furthermore, the second somatic mutation in a carrier of the constitutive mutation may appear in a greater number of cells, hence the multifocal and bilateral development of such tumors is common (4, 17).
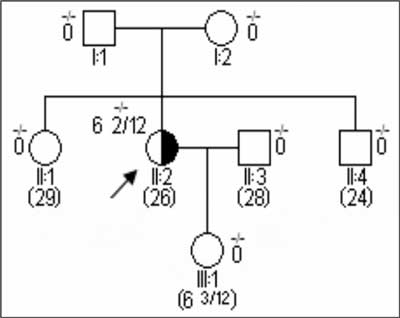
Fig. 1. Typical pedigree of a sporadic retinoblastoma without a constitutive mutation.
Legend for pedigrees presented in figures 1-4:
-/- a constitutive mutation excluded
+/- a constitutive mutation present in one allele
3 2/12 the age of diagnosis in years and months
(12) current age in years
Bilateral and unilateral multifocal cases of Rb are usually associated with the presence of a constitutive mutation which is either transmitted from ancestors or acquired de novo. In the majority of cases, they appear before the age of 3 years (fig. 2, 3). Constitutive mutations may also arise de novo in germ cells (fig. 2). However, tumors associated with such mutations have a sporadic family pedigree, they show different symptoms and an earlier onset, which make them more similar to familial cases. Certain patients, (20% in our own studies) despite the unilaterality of the Rb at the moment of diagnosis, carry a constitutive mutation. Compared to other unilateral cases of Rb, tumors in such patients occur earlier and are multifocal. What more, those patients are at a high risk of a new primary tumor growth in the same or other eye (10, 13, 14, 16, 17).
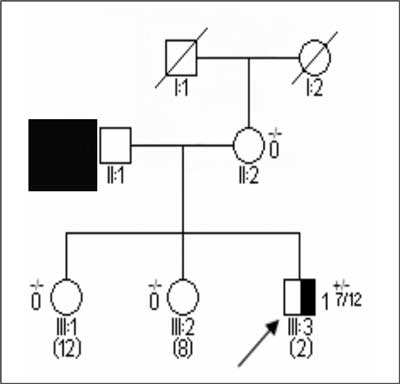
Fig. 2. Typical pedigree of a hereditary retinoblastoma as a consequence of a constitutive mutation de novo.
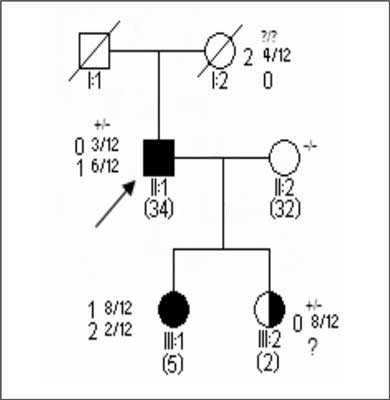
Fig. 3. Typical pedigree of hereditary retinoblastoma as a consequence of a de novo constitutive mutation ? (II-1) with complete (III-1) and possibly incomplete expression and penetrance (III-2).
RETINOBLASTOMA GENE AND PROTEIN
The RB1gene is of medium size (200 kb). It is located in the 13q14 chromosome region, and comprises a promoter and 27 exons. There is a single RNA transcript of 4.8 kb size known. Therefore, the alternative RNA splicing of the RB1gene presumably does not take place.
The RB1gene encodes a p105RB1 protein, which belongs to the pocket protein family and is a nuclear phosphoprotein of 105-110 kDa size. It participates in complex regulatory pathways, which control the pass from G1 into S phase in the cell cycle. Therefore, it is one of the key proliferation regulators. This process concerns all cells in the organism and the gene shows a high evolutionary conservation. The protein affects the functions of many other genes by inactivation (sequestration) of one of the main transcription regulators – E2F factor. This is performed by E2F binding to the pRB1 pocket domain. It disables the transactivation of dependent genes by E2F. The result is that cells remain in the G0 phase. Only the pRB1 protein with the hypophosphorylation on its serine residues has the ability for binding E2F; the hyperphosphorylation of the protein releases the transcription factor and enables the passing from G1 phase to S phase and the progression of the cell cycle. Some proteins of oncogenic viruses such as the E7 protein of the papilloma virus, the E1A protein of adenovirus, T-antigen of the SV40 virus and others bind competitively to the pocket domain of the pRB1 protein. This phenomenon allows for associating the virus ontogenesis processes with the appearance of genetically determined neoplasms. The pocket domain of pRB1 protein may bind many other proteins containing the -Leu-x-Cys-x-Glu- chain (4, 6, 7, 15, 26). Sequence mutations found in patients with Rb diagnosed are predominantly located in the protein pocket domain and its vicinity. They can affect the affinity for transcription factors. When such a blocking is not complete, mutations are of a low penetrance (see below).
The universal regulatory character of the pRB1 protein can explain its mutation involvement in arising from many others tumors, such as osteosarcoma, bladder cancer, small cell lung cancer etc., co-occurring in families with retinoblastoma history (1).
The activity of the RB1gene is regulated by the promoter and the associated transcription regulators. Few known promoter mutations downgrade this regulation and show clinical features of low penetrance (see below). One of them and the first de novo recognized mutation was identified in author's center (28, 29). Regular cell functioning requires the presence of only one normal allele. Constitutive mutation carriers, apart from having a predisposition to develop Rb, do not differ from healthy individuals having two normal alleles.
TIME COURSE AND CLINICAL PATTERN
The peak incidence of retinoblastoma is at the age of about 42 months. More than 90% of cases are diagnosed before the age of 5 years. There are known cases where the tumor was diagnosed immediately after birth. Early symptoms indicating Rb are squinting, red eye and inflammation of the eyeball. However, the tumor is usually diagnosed by symptoms appearing later on. They are exophthalmos and a "cat's eye” noticed by parents – white pupil, which comes from the caseous surface of the tumor itself. Treatment at an early stage frequently allows preservation of the affected eyeball even with the vision field worsened. The diagnosis at a later stage, unfortunately the most common situation, when the "cat's eye” sign is present, usually requires the removal of the eyeball. It is often associated with radiotherapy and chemotherapy (6, 17).
Metastases of retinoblastoma are rare. The tumor spreads usually per continuitatem, mainly into adjacent tissues via the optic nerve. The prognosis depends mainly on the quality of diagnostics and health care. In developed countries with good medical care standards the mortality rate hardly exceeds 8% of the cases, and eyeball removal is necessary in about 10% of cases. In developing countries the mortality rate can reach 100%.
MOLECULAR BACKGROUND OF THE HEREDITARY RETINOBLASTOMA
Currently RB1gene mutations have been noted in databases in ~1000 patients with constitutive retinoblastoma (8, 14, 25). They are classified with all generally known mutation categories: different types of point mutations, translocations, insertions, and deletions. Recently a relatively great occurrence of deletions among all mutations has been found. In previous years they were neglected. Particular classes of RB1gene alterations are point mutations in exonic and intronic sequences, mutations generating aberrant splicing, point mutations of promoter, and modifications of their methylation status (epigenetic changes) without alterations of sequence per se.
The majority of hereditary mutations, particularly in bilateral cases, are nonsenses and frameshifts. They generate premature stop codons and are localized in exons 1-25. No mutation has been identified until now in the last two exons 26 and 27. Although, they are potentially mutation prone sites due to two potential hot spots, CGA.
Hereditary predisposition to retinoblastoma is associated with the appearance of constitutive mutations of the RB1gene. There is a full service molecular RB1gene testing facility available in the author's institution (9, 27, 31). Gene sequence analysis is very expensive. However, it has been demonstrated that the expenses are lower after non-carriers have been excluded from the tests than the costs of extended prophylactic care for all children in the susceptible family (19). Full RB1gene sequencing identifies the mutation in about 80% of families, in which Rb is apparently hereditary. There are some preselective strategies (e.g. SSC, DGGE, PTT) that make this process easier to some extent but their sensitivity is lower than that of sequencing. Identification of the mutation in an affected individual allows for focusing the analysis on only the selected gene fragment in other members of the family (6, 8). Linkage analysis with intra-gene molecular probes can also be used especially in the diagnosis of numerous families (6, 12, 13, 29). Not only a classic PCR technique (exon-by-exon sequencing) but also multiplex PCR and quantitative multiplex PCR techniques (allowing simultaneous analysis of a number of exons), and new biochemical techniques for the detection of results of a mutation (e.g. HPLC) were introduced (21, 25, 30, 31).
Constitutive DNA extracted from a peripheral blood sample is analysed. Such samples contain encoding fragments (exons), as well as non-coding sections (introns) of the gene. The use of cDNA obtained in vitro from RNA and containing only the encoding sections of DNA improves the assessment technique and the following interpretation (8, 9). The diagnosis of the tumor DNA is not essential to identify the constitutive mutation. Nevertheless, it can verify the procedure and provide with unique information on somatic mutations ("second hit” and sporadic) (12, 13).
The findings of the mutations observed are stored in two free online databases. The Lohmann's Database holds fewer mutations and is rarely updated, but allows for simple discerning of constitutive mutations from somatic "second hits”. There is more new data (932 mutations) in the Valverde et al's Database, but the differentiation is not so simple. However, the analysis of the latter database has revolutionized the knowledge of constitutive mutations in the RB1gene (14, 25).
Until recently, it was considered that the mutations have random localization (8). It has been proven though that there are preferential sequences in the RB1gene where mutations accumulate. They correspond to the active sites of the pRB1 protein and especially to the pocket domain. 79% of the repetitive mutations are transitions C/T which appear in 11 triplets of the arginine codons (CGA); 40% of those are located in exons 8, 10, 11, 14, 15, 17, 18, and 23 (13, 25). These findings significantly change diagnostic sequencing technique, time and costs.
As expected, the immense majority of the mutations make the synthesis of the protein impossible or result in an inactive product. These are basically stop mutations (42% of cases), but also some types of deletion and splicing site mutations occur.
In contrast to the mutations that inactivate the product, the mutations that modify the activity of the molecule, yet not stopping the synthesis of the pRB protein completely, are randomly distributed.
This group consists of nonsense mutations and some of splicing site mutations. Stop codon producing mutations prevail. There is lower penetrance (60-70%) of promoter mutations and some splicing sites mutations (9, 12, 23, 28). Mutations skipping a generation are observed (obligatory carriers remain healthy) in susceptible families. Clinical presentation of the tumor is similar to that observed in Rb development as a result of somatic mutations (8, 22). The assessment of the risk of developing a tumor in such families may be difficult. However, mutations of low penetrance probably form only a small part of all mutations observed in the RB1gene (16, 18, 21, 22, 23, 24).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Abramson DH: Second Non-Ocular Cancers in Retinoblastoma: A Unified Hypothesis, Ophth Genet 1999; 20: 193- 203.
2. Albrecht P, Ansperger-Rescher B, Schuler A et al.: Spectrum of Gross Deletions and Insertions int the RB1 Gene in Patients with Retinoblastoma and Association With Phenotypic Expression. Hum Mutat 2005; 26, 5: 437-445.
3. Cowell JK, Gallie BL: Which Retinoblastoma Patients Should be Screened for RB1 Mutations? Eur J Cancer 1998; 34: 1825-1826.
4. DiCiomino D, Gallie B, Bremner R: Retinoblastoma: The disease, Gene and Protein Provide Critical Lead to Understand Cancer. Cancer Biol 2000; 10: 255-269.
5. Gallie BL, Ellsworth RM, Abramson DH et al.: Retinoma: Spontaneous Regression of Retinoblastoma or Benign Manifestation of Mutation? Br J Cancer 1982; 45: 513-521.
6. Gallie BL, Dunn JM, Chan HS et al.: The Genetics of the Retinoblastoma: Relevance to the Patient. Pediatr Clin North Am 1991; 38: 299-315.
7. Gutkind JS (Ed.): Signalling Networks and Cell Cycle Control: The Molecular Basis of Cancer and other Diseases. Humana Press, Champaing, IL, 2000.
8. Harbour WJ: Overview of RB Gene Mutations in Patients with Retinoblastoma. Implications for Clinical Genetics. Ophthalmology 1998; 105: 1442-1447.
9. Jakubowska A, Zajaczek S, Haus O et al.: "Novel RB1 Gene Constitutional Mutations Fund In Polish patients with familial and/or Bilateral Retinoblatoma.” Hum Mutat 2001; 18: 459. Online Mutations in Brief #456.
10. Kloss K, Warissch P, Greger V et al.: Characterisations of Deletions at the Retinoblastoma Locus in Patients with Bilateral Retinoblastoma. Am J Med Genet 1991; 39: 196-200.
11. Klutz M, Bockmann D, Lohmann DR: A Parent-of -Origin Effect in Two Families with Retinoblastoma is Associated with a Distinct Splice Mutation in the RB1 Gene. Am J Hum Genet 2002; 71: 174-179.
12. Lohmann DR, Brandt B, Höpping W et al.: The Spectrum of RB1 Germline Mutations in Hereditary Retinoblastoma. Am J Hum Genet 1996; 58: 940-949.
13. Lohmann DR, Gerick M, Brandt B et al.: "Constitutional RB1 Gene Mutations in Patients with Isolated Unilateral Retinoblastoma.” Am J Hum Genet 1997; 61: 282-294.
14. Lohmann DR, Brenda LG: Retinoblastoma Gene Clinics 2003: http://www.geneclinics.org/profiles/retinoblastoma/details.html
15. Lohmann DR, Galllie BL: Retinoblastoma: Revisiting the Model Prototype of Inherited Cancer. Am J Med Genet. C, Semin. Med Genet 2004; 129, 1: 23-28.
16. Munier FL, Thonney F, Girardet A et al.: "Evidence of Somatic and Germinal Mosaicism in Pseudo-Low Penetrant Hereditary Retinoblastoma, by Constitutional and Single Sperm Mutation Analysis.” Am J Hum Genet 1998; 63: 1903-1908.
17. Murphee AL, Clark RD: Retinoblastoma. [In:] Emery's and Rimoin's Principles and Practice of Medical Genetics. D.L.Rimoin, J.M.Connor, R.E.Pyeritz, B.R. Korf (eds), Churchill – Livingstone, London 2002; 3: 3604-3636.
18. National Cancer Institute, 2006. Retinoblastoma Treatment: http://www.cancer.gov/cancertopics/pdq/treatment/retinoblastoma//healthprofessional
19. Noorani HZ, Khan HN, Gallie BL et al.: Cost Comparison of Molecular vs Conventional Screening of Relatives at Risk for Retinoblastoma. Am J Hum Genet 1996; 59: 301-307.
20. Otterson GA, Modi S, Nguyen K et al.: Temperature Sensitive RB Mutations Linked to Incomplete Penetrance of Familial Retinoblastoma in 12 Families. Am J Hum Genet 1999; 65: 1040-1046.
21. Richter S, Vandezande K, Chen N et al.: Sensitive and Efficient Detection of RB1 Gene Mutations Enhances Care for Families with Retinoblastoma. Am J Hum Genet 2003; 72: 253-269
22. Scheffer H, te Meerman GJ, Kruize YC et al.: Linkage Analysis of Families with Hereditary Retinoblastoma: Nonpenetrance of Mutation. Am J Hum Genet 1989; 45: 252-260.
23. Schubert EL, Strong LC, Hansen MF: A Splicing Mutation in RB1 in Low Penetrance Retinoblastoma. Hum Genet 1997; 100: 557-563.
24. Taylor M, DeHainaut C, Desjardins L et al.: Genotype – Phenotype Correlations in Hereditary Familial Retinoblastoma. Human Mutation 2007; 28, 3: 284-293.
25. Valverde JR, Alonso J, Palacios I et al.: RB1 Gene Mutation Up-to Date: a Meta-Analysis Based on 932 Reported Mutations Available in a Searchable Database. BMC Genetics 2005; 6: 1-9, http://www.biomedcentral.com/1471-2156/6/53
26. Xiao B, Spencer J, Clements A, Ali-Khan N et al.: "Crystal Structure of the Retinoblastoma Tumor Supressor Protein Bound to E2f and the Molecular Basis of its Regulation.” Proc Natl Acad Sci USA 2003; 100.
27. Zajaczek S, Jakubowska A, Kurzawski G et al.: Age At Diagnosis to Discriminate those Patients for whom Constitutional DNA Sequencing is appropriate in Sporadic Unilateral Retinoblastoma. Eur J Cancer 1998; 34: 1919-1921
28. Zajaczek S, Jakubowska A, Górski B et al.: Frequency and Nature of Germline RB-1 Gene Mutations In a Series of Patients with Sporadic Unilateral Retinoblastoma. Eur J Cancer 1999; 35: 1824-1827.
29. Zajączek S: Ocena konstytucyjnych mutacji genu Rb-1 i niestabilności chromosomów w diagnostyce dziedzicznych postaci i badaniach patogenezy siatkówczaka jednostronnego. Praca habilitacyjna. Pomorska Akademia Medyczna w Szczecinie 1999; 1-80.
30. Zajączek S: Genetics of Retinoblastoma in Clinical Practice. Ann Diagn Paed Pathol 2003; 3: 35-39.
31. Zajączek S: Genetyka siatkówczaka w praktyce klinicznej – przegląd nowych zagadnień. Okulistyka 2007; 10: 7-13.
