© Borgis - Postępy Nauk Medycznych 11/2010, s. 870-876
*Aleksandra Tołoczko-Grabarek, Joanna Trubicka, Jan Lubiński
Clinical genetics of kidney cancer
Genetyka kliniczna nowotworów nerek
International Hereditary Cancer Centre, Department of Genetics and Pathology, Pomeranian Medical University, Szczecin, Poland
Head of the Genetics and Pathology Unit: prof. dr hab. med. Jan Lubiński
Streszczenie
Wśród nowotworów nerki można wyodrębnić 2 główne grupy:
1. Guz Wilmsa – nephroblastoma.
2. Raki nerki – adenocarcinoma (carcinoma clarocellulare, carcinoma papillare, colecting duct carcinoma, chromophobic cell carcinoma), carcinoma urotheliale.
Postać rodzinna guza Wilmsa stanowi 1-2% tych guzów.
Dotychczas opisano 14 genetycznie uwarunkowanych zespołów związanych ze zwiększonym ryzykiem guza Wilmsa. Mutacje konstytucyjne będące przyczyną powstawania dziedzicznego nephroblastoma dotyczą najczęściej jednego z czterech genów: WT1, WT2, FWT1, FWT2. Ryzyko zachorowania na guza Wilmsa u nosicieli tych mutacji wynosi 5-90% w zależności od zmutowanego genu.
Opisano 19 zespołów dziedzicznej predyspozycji do nowotworów, w przebiegu, których w nerkach mogą powstać rak jasnokomórkowy (CCRC), rak brodawkowaty (PRCC) lub rak z nabłonka przejściowego. Najczęściej rozpoznawanym zespołem jest rodzinny rak jasnokomórkowy nerki (F-CCRC). Wśród wszystkich rodzin CCRC 5% spełnia kryteria definitywne F-CCRC, zaś 13% stanowią rodziny podejrzane o F-CCRC.
W większości przypadków F-CCRC nie znaleziono mutacji będących przyczyną agregacji CCRC w rodzinach. W naszych Ośrodku wykazaliśmy, że istotną przyczyną powstawania CCRC jest konstytucyjna zmiana w genie CHEK2. Nadal jednak kluczową rolę w rozpoznawaniu F-CCRC odgrywa analiza rodowodowo-kliniczna. W naszym Ośrodku w rodzinach z pojedynczym CCRC wysuwamy podejrzenie F-CCRC stosując jako kryteria:
a. zdiagnozowanie CCRC poniżej 55. r.ż. lub
b. wystąpienie raka żołądka lub płuca u krewnych I stopnia pacjenta z CCRC.
U osób z rodzin z rozpoznanym F-CCRC stosujemy schemat badań kontrolnych pozwalający na wczesne wykrycie raka nerki, jednak postepowanie lecznicze nie jest ściśle określone.
Ewentualne schematy leczenia będą mogły być ustalone po analizie przebiegu klinicznego leczonych w różny sposób dużych grup raków z rodzin z F-CCRC.
Summary
Among kidney malignancies two main groups can be distinguished:
1. Wilms' tumor – nephroblastoma.
2. Kidney cancer – adenocarcinoma (carcinoma clarocellulare – CCRC, carcinoma papillare – PRCC, collecting duct carcinoma, chromophobe cell carcinoma), carcinoma urotheliale.
Wilms' tumor and kidney cancer can develop as a result of strong hereditary predisposition.
Familial Wilms' tumor constitutes 1-2% of all cases.
Fourteen different syndromes with increased risks of Wilms' tumor have been reported. Hereditary Wilms' tumor is caused by constitutional mutation in WT1, WT2, FWT1 or FWT2 genes. Depending on the mutated gene, the risk of Wilms' tumor varies between 5 and 90%.
Kidney cancer can appear in 19 different hereditary cancer syndromes. One of the most often diagnosed is Familial Clear Cell Renal Cancer Syndrome (F-CCRC). Based on our results, in 5% of cases we can diagnose F-CCRC, and 13% are families with only one affected by CCRC but with a high probability of CCRC in their relatives.
We found that mutations in CHEK2 are associated with an increased risk of CCRC, but still the pedigree and clinical data are crucial for appropriate diagnosis.
The results of our study of CCRC patients revealed that F-CCRC syndrome could be recognized if:
a. CCRC was diagnosed before the age of 55 years.
b. Ist degree relatives of CCRC patient were affected by stomach or lung cancers.
In all individuals with recognized F-CCRC syndrome, specific surveillance is recommended.
Medical treatment for CCRC patients with F-CCRC is not defined yet and should be determined.

WILMS' TUMOR
Wilms' tumor ( nephroblastoma) is the most common malignant renal neoplasm in children. It is diagnosed with the frequency of 1:10,000 children under the age of 15 years (1). The majority of cases result from de novo mutations and is an isolated case within the family. Familial cases of Wilms' tumor constitute 1-2% of all cases.
Genetic basis for Wilms' tumor
Constitutive mutations leading to hereditary nephroblastoma include mainly one of four genes: WT1, WT2, FWT1, FWT2 (2, 3). In approximately 10% of patients with nephroblastoma the condition is associated with cryptorchidism, hypospadias, facial dysmorphy or developmental defects syndromes: BWS, WAGR. DDS (tab. 1).
Table 1. Genetic syndromes associated with high risk of Wilms' tumor.
| Syndrome | Phenotype | Gene/locus | Inheritance | Wilms' tumor risk |
| Familial aggregation of Wilms' tumors without other clinical pathologies | Familial aggregation of Wilms' tumors; more frequently unilateral tumors. | WT1(11p13)
WT2 (11p15)
FWT1(17q)
FWT2(19q) | AD | WT1, WT2 genes mutations ~25%; FWT1 gene mutation - 30%; FWT2 gene mutation 70% |
| Denys-Drash syndrome (DDS) | Wilms' tumor, glomerulopathy, pseudohermaphroditism. | Point mutation, WT1 (11p13) | Germinal de novo mutation, rare family aggregation, AD | 90% |
| WAGR | Wilms' tumor, aniridia, genitourinary anomalies, mental retardation. | Deletion in WT1(11p13) | Germinal de novo mutation, rare family aggregation, AD | 30% |
| Beckwith-Wiedemann Syndrome (BWS) | Macrosomia, macroglossia, midline abdominal wall defects
Other findings: visceromegalia, hipoglycemia, hemihypertrophy, genitourinary anomalies, embryonic tumors. | Locus WT2 (11p15) | Germinal de novo mutation, 15% of cases are familial BWS, AD | 5% |
| Simpson-Golabi-Behmel (SGB) | Gigantism, developmental defects, embryonic tumors. | GPC3 (Xq26) | XR | High in boys, undetermined |
| HPT-JT (hyperparathyroidism-jaw tumor) | Adenomas, rarely parathyroid cancer in early years, osteofibrous mandible tumors, Wilms' tumor | HRPT2 (1q21-q31) | AD | High, undetermined |
| Perlman | Macrosomia, visceromegalia, facial dysmorphies, cryptorchidism | ? | AR | < 25% in siblings |
| Trisomy 18 | Multiple organ defects, mental retardation, Wilms' tumor | | | Low, undetermined |
| Isolated hemihypertrophy | Hemihypertrophy | ? | ? | <5% |
| Aniridia | Aniridia | PAX6 | ? | 1.5% |
| Breast-ovarian cancer | Breast and ovarian cancer | BRCA1 (17q11), BRCAX | AD | Low |
| Li-Fraumeni | Sarcoma, leukemias, brain tumors, breast cancer | p53 (17p13) | AD | Low |
| Neurofibromatosis-type 1 | Neurofibromas, café-au-lait spots, Lisch nodules, optic glioma | NF1 (17q11) | AD | Low |
| Bloom | Short stature, facial rash, leukemias, lymphomas, colorectal cancer and other cancers | BLM (15q26) | | Low |
Wilms' tumor has autosomal dominant pattern of inheritance with incomplete penetrance.
Hereditary nephroblastoma with the above mentioned mutations develop bilaterally more frequently than in sporadic cases (approx. 20% vs. 5%) (1).
Diagnostics principles of the genetic factors for Wilms' tumor
Genetic predisposition for Wilms' tumor is rarely diagnosed as it is underestimated to:
1) take the medical history of distant relatives necessary for familial nephroblastoma detection;
2) link the occurrence of nephroblastoma with the aggregation of other neoplasms, e.g. breast and ovarian cancer or neuromas and sarcomas in families with NF1 mutation;
3) evaluate completely the dysmorphies that enable seeing a genetic predisposition for Wilms' tumor associated with developmental defects.
Like in cases of other familial hereditary cancer syndromes Wilms' tumor diagnosis requires the following steps:
– In-depth analysis of familial medical history data:
a) Wilms' tumor incidence in first- and second-degree relatives, as well as third- and forth-degree;
b) Familial aggregation of other neoplasms;
– Physical examination;
– Cytogenetic assay;
– Molecular DNA assay (4).
Cytogenetic assay is particularly useful when detecting hereditary Wilms' tumors arising due to de novo mutations. It is relatively inexpensive and widely available compared to molecular DNA assay. It was shown that karyotyping is effective in identifying translocations in locus WT2 in patients with BWS, while in situ, hybridization is a sensitive method of detection of large deletions in WT1 gene in people with WAGR syndrome.
In molecular labs conducting scientific research, the complete sequence analysis of WT1, p53 and GPC3 is available enabling point mutation detection. Unfortunately, the remaining genes responsible for hereditary nephroblastoma (WT2, FWT1 and FWT2) have not been cloned yet.
Screening in families with high risk of Wilms' tumor
In families predisposed to Wilms' tumor abdominal ultrasounds should be performed every 3 months from childbirth to the age of 8 years, subsequently every 6 months until 12 years of age (and less frequently afterwards) (1, 5). In cases of unclear lesions or nephrogenic tissue presence (there is some opinions of nephrogenic tissue origin of nephroblastoma), MRI or CT is indicated (1, 6). In Beckwith-Wiedemann Syndrome (BWS) because of the increased risk of other types of cancer the screening should additionally include AFP (alpha-fetoprotein) testing for hepatoblastoma detection. Certain authors recommend performing periodical chest X-rays and VMA (Vanillylmandelic acid) urinalysis test for early neuroblastoma identification (1).
RENAL CELL CARCINOMA (RCC)
Renal cell carcinoma constitutes about 3% of all adult malignancies. In Poland 2,000 new cases are detected every year (7). Clear Cell Renal Carcinoma (CCRC) represents approximately 80% of all diagnosed renal cell carcinomas (7). 1-2% of all RCC is linked with high genetic risk (8).
Thus far 19 syndromes with hereditary cancer predispositions have been described that may lead to the following kidney neoplasms: CCRC, Papillary Renal Cell Carcinoma (PRCC) or collecting duct carcinoma.
Clear Cell Renal Carcinoma (CCRC)
The best known hereditary cancer predisposition syndrome that may lead to CCRC is Von Hippel-Lindau (VHL) disease presented in other chapters of this book (7, 9, 10, 11, 12).
Familial Clear Cell Renal Carcinoma is the most frequently diagnosed syndrome (F-CCRC), in the course of which at least 2 cases of CCRC and the lack of VHL syndrome features are stated (8, 13, 14, 15).
According to our data, the CCRC definitive criteria are met in 5% of all families with CCRC, while in 13% of families there is one case of CCRC with a high risk of subsequent CRCC development.
A genetic predisposition to cancer can be diagnosed more precisely if a constitutive mutation in one of the genes responsible for cancer development is detected. Owing to the availability of mutation analysis in BRCA1, BRCA2, MSH2, or MLH1 genes, an extremely efficient diagnosis of predisposition to breast, ovarian, colorectal and endometrial cancer can be reached.
The gene responsible for F-CCRC occurrence has not been identified yet. In some families' cases cytogenetic assay is helpful in F-CCRC diagnosis.
Currently, there are reports of 3 families with F-CCRC development linked with constitutive translocations. In 1979 Cohen et al. presented familial reciprocal translocation between chromosome 3 and 8 [t(3;8) (p14,2;q24,1)] giving rise to bilateral CCRC at a young age (between 37 and 59) (16). One female mutation carrier had multifocal papillary and anaplastic thyroid cancer. Gemmill et al. in 1998 suggested that t(3;8) leads to the fusion of FHIT and TRC8 genes and probably produces an increasing renal cancer risk. In 1988 Kovacs published a paper presenting a family with constitutive translocation t(3;6) (p13;q25,1). A male carrier of this translocation at the age of 53 years developed bilateral, multifocal CCRC (17, 18).
Another case of familial renal carcinoma caused by translocation was reported in 1998 by the Koolen and Bodmer team (19, 20). In four carriers of reciprocal balanced translocation t(2;3) (q35;q21) RCC developed at the age of 40, 53, 54 and 68 years, including 3 cases of CCRC. One of the carriers had bladder squamous cell carcinoma.
Identical translocation was identified in 1996, in our center, in two brothers with bilateral CCRC, whose 5 other relatives died of renal carcinoma (21).
Van Kessel et al. tested 57 carriers of various reciprocal translocations of chromosome 3 from 10 families. Since four subjects had renal carcinoma a conclusion was drawn that chromosome 3 translocation carriers had an increased risk of renal carcinoma, particularly if the breakage site is located near the centromere (22). The condition develops at an early age (ca. 45), it is multifocal and bilateral.
In the majority of F-CCRC cases presented in literature, an underlying mutation was not found (23, 24, 25, 26, 27, 28). Recently, in our center, it has been shown that constitutive mutation I157T in CHEK2 gene is responsible for CCRC development (29).
However patient history analysis remains the basic method for F-CCRC detection.
Our team defined patient history and clinical symptoms-based criteria resting on first-degree relatives evaluation ("nuclear pedigree” criteria) as enabling the detection of F-CCRC families despite no cases of CCRC in first-degree relatives.
Based on our studies, we proved that in families with a single case of CCRC, F-CCRC can be suspected (fig. 1) when:
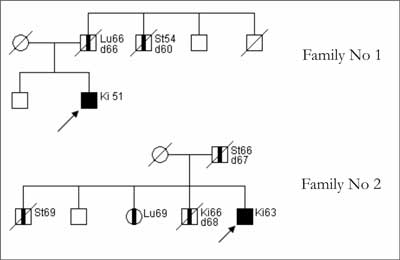
Fig. 1. Pedigree charts for families with F-CCRC.
– CCRC is diagnosed below the age of 55 years, or
– gastric or lung cancer occurred in first-degree relatives of the patient with CCRC.
Screening in families with F-CCRC
The characteristic trait of the CCRC condition is its asymptomatic initial period. Clinical manifestations appear along with disease progress. Data from published studies suggests that proper medical management of families with F-CCRC may decrease the risk of developing cancer (25, 30, 31).
However, there are no verified screening programs for early F-CCRC detection.
Levinson suggests that members of a family with F-CCRC should have kidney ultrasounds every 2-3 years starting at the age of 30 years (22). The scheme is accepted by other authors (2, 32), however, it is admitted arbitrarily.
Our analysis suggests that the commencement of screening the members of a family with F-CCRC should depend on when the first appearanceof the disease in the family occured and precede the earliest detection of RCC by 15-20 years but should not start later than at the age of 40 years.
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Zajączek S, Dębniak T, Lubiński J et al.: Nowotwory dziedziczne u dzieci. Guz Wilmsa. Wsp Onk 2002; 6: 300-7.
2. Dome JS, Coppes MJ: Recent advances in Wilms tumor genetics. Current opinion in pediatrics 2002; 14: 5-11.
3. Ruteshouser EC, Huff V: Familial Wilms Tumor. Am J Med Genet 2004; 129: 29-34.
4. Zajączek S, Lubiński J: Zasady poradnictwa genetycznego u rodzino podwyższonym ryzyku choroby nowotworowej. Nowotwory 1999; 49: 71-2.
5. Little M, Wells C: A clinical overview of WT1 gene mutations. Human Mutat 1997; 9: 209-25.
6. Borer JG, Kaefer M, Barnewolt CE et al.: Renal findings on radiological followup of patients with Beckwith-Wiedemann syndrome. J Urol 1999; 161: 235-9.
7. Borkowski A, Czaplicki M: Nowotwory i torbiele nerek. Wyd 1. PZWL, 2002.
8. Goldman SM, Fishman EK, Abeshouse G et al.: Renal cell carcinoma diagnosed in three generations of a single family. South Med J 1979; 72: 1457-9.
9. Krzystolik K, Cybulski C, Lubiński J et al.: Wczesna diagnostyka bezobjawowych raków nerek w rodzinach z zespołem von Hippel-Lindau w Polsce. Urol Pol 1998; 51: 171-81.
10. Neumann HP: Basic criteria for clinical diagnosis and genetic counseling in von Hippel-Lindau syndrome. J Vasc Dis 1987; 16: 220-6.
11. Neumann HP, Bender BU, Berger DP et al.: Prevalence, morphology and biology of renal cell carcinoma in von Hippel-Lindau disease compared to sporadic renal cell carcinoma. J Urol 1998; 160: 1248-54.
12. Neumann HPH, Zbar B: Renal cysts, renal cancer and von Hippel-Lindau disease. Kidney Int 1997; 51: 16-26.
13. Erlandsson R, Boldog F, Sümegi J et al.: Do human renal cell carcinomas arise by a double-loss mechanism? Cancer Genet Cytogenet 1988; 36: 197- 202.
14. Franksson C, Bergstrand A, Ljungdahl I et al.: Renal carcinoma (hypernephroma) occurring in 5 siblings. J Urol 1972; 108: 58-61.
15. Reddy ER: Bilateral renal cell carcinoma – unusual occurrence in three members of one family. Br J Radiol 1981; 54: 8-11.
16. Cohen AJ, Li FP, Berg S et al.: Hereditary renal-cell carcinoma associated with a chromosomal translocation. N Engl J Med 1979; 301: 592-5.
17. van Kessel AG, Wijnhoven H, Bodmer D et al.: Renal cell cancer: chromosome 3 translocations as risk factor. J Nat Cancer Inst 1999; 91: 1159-60.
18. Maher ER, Yates JRW: Familial renal cell carcinoma: clinical and molecular genetic aspects. Br J Cancer 1991; 63: 176-9.
19. Bodmer D, Eleveld MJ, Ligtenberg MJ et al.: An alternative route for multistep tumorigenesis in a novel case of hereditary renal cell cancer and t(2:3) (q35;q21) chromosome translocation. Am J Hum Genet 1998; 62: 1475-83.
20. Koolen MI, van der Meyden AP, Bodmer D et al.: A familial case of renal cell carcinoma and a t(2;3) chromosome translocation. Kidney Int 1998; 53: 273-5.
21. Borówka A, Zajączek S: Rodzinne występowanie raka jasnokomórkowego nerki. Doniesienie zjazdowe: 26 Kongres PTU, Poznań 1996.
22. Kovacs G, Brusa P, De Rirse W: Tissue-specific expression of a constitutional 3;6 translocation: development of multiple bilateral renal-cell carcinomas. Int J Cancer 1989; 43: 422-7.
23. Levinson AK, Johnson DE, Strong LC et al.: Familial renal carcinoma: hereditary or coincidental? J Urol 1990; 144: 849-51.
24. Li FP, Marchetto DJ, Brown RS: Familial renal carcinoma. Cancer Genet Cytogenet 1982; 7: 271-5.
25. Teh B, Giraud S, Sari F: Familial non- VHL non-papillary clear-cell renal cancer. Lancet 1997; 349: 848-9.
26. Woodward ER, Clifford SC, Astuti D et al.: Familial clear cell renal carcinoma (FCRC): clinical features and mutation analysis of the VHL, MET, and CUL2 candidate genes. J Med Genet 2000; 37: 348-53.
27. Woodward ER: Familial non-syndromic clear cell renal cell carcinoma. Current molecular medicine 2004; 4 (8): 843-8.
28. Woodward ER, Ricketts C, Killick P et al.: Familial Non-VHL Clear Cell (Conventional) Renal Cell Carcinoma: Clinical Features, Segregation Analysis, and Mutation Analysis of FLCN. Clinical cancer research 2008; 14 (18): 5925-30.
29. Cybulski C, Górski B, Huzarski T et al.: CHEK2 is the multiorgan cancer susceptibility gene. Am J Hum Genet 2004; 75: 1131-5.
30. Herring JC, Enquist EG, Chernoff A et al.: Parenchymal sparing surgery in patients with hereditary renal cell carcinoma: a 10-years experience. J Urol 2001; 165: 777-81.
31. Schmidt L, Junker K, Weirich G et al.: Two North American families with hereditary papillary renal carcinoma and identical novel mutations in the MET proto-oncogene. Cancer Res 1998; 58: 1719-22.
32. Walther MM, Choyke PL, Glenn G et al.: Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol 1999; 161: 1475-9.
33. Bosniak MA, Rofsky NM: Problems in the detection and characterization of small renal masses. Radiology 1996; 198: 638-41.
34. Choyke PL, Filling-Katz MR, Shawker TH et al: Linehan WM. von Hippel-lindau disease: radiologic screening for visceral manifestation. Radiology 1990; 174: 815-20.
35. Curry NS: Small renal masses (lesions smaller than 3 cm): imaging evaluation and management. Am J Radiol 1995; 164: 355-62.
36. Jamis-Dow CA, Choyke PL, Jennings SB et al.: Small (
37. Lightfoot N, Conlon M, Kreiger N et al.: Impact of noninvasive imaging on increased incidental detection of renal cell carcinoma. Eur Urol 2000; 37: 521-7.
38. Shinohara N, Nonomura K, Harabayashi T et al.: Nephron sparing surgery for renal cell carcinoma in VHL disease. J Urol 1995; 154: 2016-9.
39. Walther MM, Linehan WM: Nephron sparing surgery for renal cell carcinoma in von Hippel-Lindau disease. J Urol 1996; 156: 480-1.
40. Zbar B, Lerman M: Inherited carcinomas of the kidney. Adv Cancer Res 1998; 75: 163-201.
41. Zbar B, Glenn G, Lubensky I et al.: Hereditary papillary renal cell carcinoma: clinical studies in 10 families. J Urol 1995; 53: 907-12.
42. Zbar B, Tory K, Merino M et al.: Hereditary papillary renal cell carcinoma. J Urol 1994; 151: 561-6.
43. Schmidt L, Duh FM, Chen F et al.: Germline and somatic mutations in tyrosine kinase domain of MET proto-oncogene in papillary renal carcinomas. Nat Gen 1997; 16: 68-73.
otrzymano: 2010-10-01
zaakceptowano do druku: 2010-10-29
Adres do korespondencji:
*Aleksandra Tołoczko-Grabarek
International Hereditary Cancer Centre Department of Genetics and Pathology Pomeranian Medical University
ul. Połabska 4, 70-115 Szczecin
tel.: (91) 466-15-32
e-mail: otjg@interia.pl
Postępy Nauk Medycznych 11/2010Strona internetowa
czasopisma Postępy Nauk MedycznychPozostałe artykuły z numeru 11/2010:
