© Borgis - Nowa Stomatologia 3/2011, s. 134-138
*Iwona Sobiech1, Anna Grzybowska1, Elżbieta Jelonek3, Julian Komarnitki2, Dorota Olczak-Kowalczyk1
Wrodzona łamliwość kości w aspekcie stomatologicznym – ocena kliniczna w oparciu o piśmiennictwo
The inherent bone brittleness in dental aspect – clinical assessment founded on literature
1Zakład Stomatologii Dziecięcej Warszawskiego Uniwersytetu Medycznego
Kierownik Zakładu: dr hab. med. Dorota Olczak-Kowalczyk
2Zakład Anatomii Prawidłowej i Klinicznej Warszawskiego Uniwersytetu Medycznego
Kierownik Zakładu: prof. dr hab. med. Bogdan Ciszek
3Klinika Rehabilitacji Pediatrycznej Instytutu Pediatrii Centrum Zdrowia Dziecka
Kierownik Kliniki: lek. med. Anna Łukaszewska
Summary
Osteogenesis imperfecta is genetically determined disorder of connective tissue inherited in an autosomal dominant, less often recessive. Belongs to the same group of disease entities as team Ehlers’a-Dalnos’a, Marfan’a team, the team Hurler’a, pseudoxantoma elasticum. Pathology is related to mutation in genes COL1 and COL2, which encode chains alfa1 and collagen type alfa2 and, the main whites of proteins of ectodermal structural tissues. General symptoms of bone structures and irregularities of mineralized tooth tissues were discussed based on the literature. Literature reports of 150 to 250 different mutations responsible for the formation of collagen OI. Among the different classifications currently the most widely used is the division of the 4 types of OI by Silence’a, taking into account clinical and genetic aspects. The complex treatment is indispensable in patient with diagnosed inherent bone brittleness disease. Patients with known congenital fragility need a comprehensive and multi-general and dental treatments. Currently in OI from infancy for the prevention of fractures of the number and severity of drug therapy is implemented which promotes better bone remodeling. Also take into account the method of rehabilitation. Patients regardless of the classification for any of the types of OI have to be individually diagnosed. Many doctors of different specialities cooperate to try to assure the patients about the optimal development and the progress in the orthopedic and pharmacology scope and they give a chance on independent functioning in mature years by new forms of rehabilitations.

Osteogenesis imperfecta (OI), czyli wrodzona łamliwość kości została opisana ponad 200 lat temu – w 1788 r. przez Ekmana (1). Zaliczana jest do tej samej grupy jednostek chorobowych co zespół Ehlersa-Dalnosa, zespół Marfana, zespół Hurlera, pseudoxantoma elasticum (2). OI jest genetycznie uwarunkowanym zaburzeniem dotyczącym tkanki łącznej, dziedziczonym w sposób autosomalny dominujący, rzadziej recesywny (3). U podłoża większości przypadków (ok. 90%) leży mutacja w genach COL1A1 chromosomu 17q21.3-q22 i COL1A2 chromosomu 7q22.1, odpowiedzialnych za kodowanie łańcuchów polipeptydowych alfa1 i alfa2 kolagenu typu I. Piśmiennictwo donosi o 150 do 250 różnych mutacjach kolagenu odpowiedzialnych za powstawanie OI (2, 4, 5). Defekty w pierwszym typie kolagenu determinują łamliwość kości (1, 6-8). Częstość występowania zespołu wynosi 1:35000 urodzeń (3) lub jak podają inne źródła 1:20000-30000, a nawet 1: 50000 urodzeń (4, 9, 10). Wśród różnych klasyfikacji obecnie najczęściej stosowany jest podział OI na 4 typy wg Silence’a, uwzględniający objawy kliniczne i aspekty genetyczne (11).
Typ I A i B (łagodny) najczęściej występujący, dziedziczony w sposób autosomalny dominujący, powstający w wyniku mutacji genu kodującego prokolagen – COL1A1, rzadziej COL1A2. W tym typie OI struktura kolagenu jest prawidłowa, zmniejszona natomiast jest jego ilość – nawet do 50% (14). Charakterystyczne objawy ogólne to: wzrost pacjentów zbliżony do prawidłowego, umiarkowana łamliwość kości, bez deformacji lub z niewielką ich deformacją, wiotkość stawów i więzadeł, upośledzenie słuchu oraz niebieskie twardówki (6, 11) (ryc. 1). W badaniu radiologicznym widoczne zaburzona mineralizacja szkieletu oraz złamania kości z nadmierną kostniną w miejscu ich gojenia (14). W tym typie opisywane są również zaburzenia kardiologiczne w postaci poszerzenia aorty, wypadania płatków zastawki dwudzielnej oraz rzadziej spotykanego tętniakowatego rozszerzenia naczyń wieńcowych (2). Podział na 2 podtypy A i B uzależniony jest od stanu tkanek zmineralizowanych zębów. W podtypie A zęby mają prawidłową budowę (ryc. 2, 3), w podtypie B przyjmują cechy dentinogenesis imperfecta (ryc. 4) (opis poniżej) (11-14).
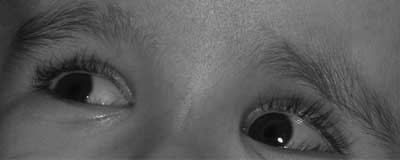
Ryc. 1. Pacjent M.R. lat 3. Osteogenesis imperfecta typ I podtyp B, charakterystyczne zabarwienie twardówek.
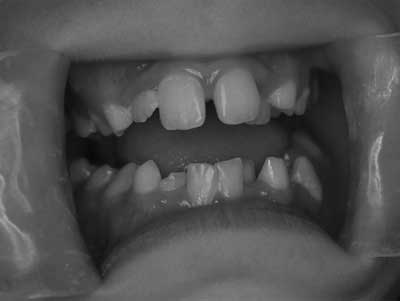
Ryc. 2. Pacjent S.Z. lat 10. Osteogenesis imperfecta typ I podtyp A. Uzębienie mieszane prawidłowe – bez cech Dentinogenesis imperfecta. Zaburzenia zgryzowe, stłoczenia.

Ryc. 3. Pacjent S.Z. lat 10. Osteogenesis imperfecta typ I podtyp A. Pantomogram – brak cech typowych dla Dentinogenesis imperfecta. Uzębienie prawidłowe.

Ryc. 4. Pacjent M.R. lat 3. Osteogenesis imperfecta typ I podtyp B. Uzębienie mleczne – specyficzne zabarwienie koron z charakterystyczną opalizacją – cechy Dentinogesis imperfecta.
Typ II (letalny perinatalny) dziedziczony heterogennie, autosomalnie dominująco (nowe mutacje) lub recesywnie. Jest najcięższą odmianą tej choroby, charakteryzującą się wysoką śmiertelnością – 50% dzieci rodzi się martwych, pozostała część umiera po urodzeniu w wyniku zaburzeń oddechowych związanych z nieprawidłową budową klatki piersiowej (4, 15, 16). Do objawów ogólnych należy zaliczyć również: znaczną łamliwość kości, deformacje, skrócenie i poszerzenie kości długich zwłaszcza kończyn dolnych, ścieńczenie skóry z tendencją do bliznowacenia, ciemnoniebieskie zabarwienie twardówek. W obrazie radiologicznym widoczne nieprawidłowa budowa żeber z licznymi zgrubieniami oraz niewłaściwe uwapnienie kości czaszki (obecność kostek wtrąconych, tzw. wromiany) (14, 15). W tym typie OI występuje zaburzenie ilościowe i jakościowe kolagenu typu I. Część autorów donosi również o spadku kolagenu w środkowej i zewnętrznej warstwie ściany naczyń myocardium (2).
Typ III A i B (progresywny deformujący) dziedziczony heterogennie, autosomalnie dominująco (75%) i recesywnie (25%) (4, 14, 16). Zaburzenie dotyczy niemożności wbudowania łańcuchów pro-α2 do cząsteczki prokolagenu, przez co prokolagen składa się wyłącznie z cząsteczek pro-α1 (14, 15). Charakterystycznymi objawami tego typu są: mnogie złamania śródmaciczne i okołoporodowe. W późniejszym wieku: niski wzrost, duże zmiany w szkielecie, znaczne i postępujące z wiekiem deformacje kości, upośledzenie słuchu, niebieskie zabarwienie twardówek ustępujące z wiekiem (14, 15). W badaniu radiologicznym stwierdza się: osteoporozę, deformacje szkieletu, skośne ustawienie żeber, nieprawidłową budowę miednicy oraz mnogie złamania kości długich. Obraz kręgosłupa opisywany jest często jako „kręgi rybie”, z licznymi nadłamaniami brzeżnymi (14, 15). W III typie OI wyróżnia się podtyp A z prawidłowym uzębieniem oraz podtyp B z zębami o cechach dentinogenesis imperfecta (14, 17).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Superti-Furga A, Pistone F, Romano C, Steinmann B: Clinical variability of osteogenesis to COL1A2 and associated with a imperfecta linked structural defect in the type I collagen molecule. Journal of Medical Genetics 1989; 26: 358-362. 2. Eskola MJ, Niemelä KO, Kuusinen PR, Tarkka MR: Interactive cardio vascular and thoracic surgery. Interact Cardio Vasc Surg 2002; 1: 83-85. 3. Szpringer-Nodzak M, Wochna-Sobańska M: Stomatologia wieku rozwojowego. Wydawnictwo Lekarskie PZWL, Warszawa 2003; 176-512. 4. Tsai C-L, Lin Y-T: Dentinogenesis Imperfecta Associated with Osteogenesis Imperfecta: Report of Two Cases. Chang Gung Med J 2003; 26 (2). 5. Alanay Y, Avaygan H, Camacho N et al.: Mutation in the gene encoding the RER protein FKBP 65 cause autosomal-recessive osteogenesis imperfecta. The American Journal of Human Genetics 2010; 86: 551-559. 6. Benušien E, Kuéinskas V: COL1A1 mutation analysis in Lithuanian patients with osteogenesis imperfecta. J Appl Genet 2003; 44 (1): 95-102. 7. Christiansen HE, Schwarze U, Pyott SM et al.: Homozygosity for a Missense Mutation in SERPINH1, which Encodes the Collagen Chaperone Protein HSP47, Results in Severe Recessive Osteogenesis Imperfecta. The American Journal of Human Genetics 2010; 86: 389-398, March 12. 8. Gautieri A, Uzel S, Vesentini S et al.: Molecular and mesoscale mechanisms of osteogenesis imperfecta disease in collagen fibrils. Med Oral Patol Oral Cir Bucal 2009; 97: 857-865. 9. Schwartz S, Joseph C, Iera D, Vu D: Biphosphonate, osteonecrosis, osteogenesis imperfecta and dental extracions: a case series. JCDA 2008; 74 (6): 537-542. 10. Lygidakis N, Smith R, Dulis C: Scanning elektron microscope of teeth in osteogenesis imperfecta type 1. Oral Surg, Oral Med, Oral Path 1996; 81,5: 567-572. 11. Pawlicki R, Knychalska-Karwan Z, Jakób-Doleżał K, Lejman T: Osteogenesis imperfecta – badania zębów mlecznych w mikroskopie optycznym,elektronowym skaningowym oraz mikroanalizatorze rtg. Czas Stomat 1998, LI, 7: 433-440. 12. Gupte T, Iyer V, Damle SG et al.: Osteogenesis imperfecta. J Indian Soc Pedod Prev Dent Special issue 2006; 44-46. 13. Gallego L, Junquera L, PelazA, Costolla S: Pathological mandibular fracture after simple molar extraction in a patient with osteogenesis imperfecta treated with alendronate. Med Oral Patol Oral Cir Bucal 2010; 1-3. 14. Sołtysiuk I, Marczuk-Kolada G: Osteogenesis imperfecta – podstawy diagnostyczne i możliwości leczenia stomatologicznego. Nowa Stomatologia 2003; 4: 168-174. 15. Adamowicz-Klepalska B, Olejniczak M: Osteogenesis imperfecta – problem interdyscyplinarny. Czas Stomat 1998; LI, 4: 241-245. 16. Sillence D: Osteogenesis imperfecta an expanding of variants. Clin Orthopat Related Res 1981; 159: 11-25. 17. Harabasz A, Latos-Bieleńska A: Osteogenesis imperfecta – obraz kliniczny i defekt molekularny. Nowiny Lek 1997; 66 (2): 175-182. 18. Waltimo J, Djanotko-Harr A, Lukinmaa P: Mild forms of dentinogenesis imperfecta in association with osteogenesis imperfecta as characterized by light and transmission electron microscopy. J Oral Pathol Med 1996; 25, 5: 256-264. 19. Lee DY, Cho T-J, Choi IH et al.: Clinical and radiological manifestations of osteogenesis imperfecta type V. J Korean Med Sci 2006; 21: 709-14. 20. Levin S: The dentition in the osteogenesis imperfecta syndromes. Clin Ortopad Related Res 1981; 159, 9: 64-74. 21. Kamboj M, Chandra A: Dentinogenesis imperfect type II: an affected family saga. Journal of oral Science 2007; 49 (3): 241-244. 22. Subramaniam P, Mathew S, Sugnani SN: Dentinogenesis imperfecta: a case report. J Indian Soc Pedod 2008; 85-87. 23. Acevedo AC, Santos LM, Dong PJ, MacDougall M: Phenotype Characterization and DSPP Mutational Analysis of Three Brazilian Dentinogenesis Imperfecta Type II Families. Cells Tissues Organs 2009; 189: 230-236. 24. Witkop CJ: Hereditary defects in enamel and dentin. Acta Genet 1957; 7: 236-239. 25. Thotakura SR, Mah T, Srinivasan R, et al.: The Non-collagenous Dentin Matrix Proteins are Involved in Dentinogenesis Imperfecta Type II (DGI-II). J Dent Res 2000; 79 (3): 835-839. 26. Shields ED, Bixler D, el-Kafrawy AM: A proposed classification for heritable human dentine defects with a description of a new entity. Arch Oral Biol 1973; 18: 543-553. 27. Kocher MS, Shapiro F: Osteogenesis imperfecta. J Am Acad Orthop Surg 1998; 6: 225-236. 28. Lee S-K, Hu JC-C, Lee K-E et al.: A Dentin Sialophosphoprotein Mutation That Partially Disrupts a Splice Acceptor Site Causes Type II Dentin Dysplasia. J Endod 2008; 34 (12): 1470-1473. 29. Ball SP, Cook PJ, Mars M, Buckton KE: Linkage between dentinogenesis imperfecta and Gc. Ann Hum Genet 1982; 46: 35-40. 30. Hirst KL, Simmons D, Feng J et al.: Elucidation of the sequence and the genomic organization of the human dentin matrix acidic phosphoprotein 1 (DMPl) gene: exclusion of the locus from the causative role in the pathogenesis of dentinogenesis imperfecta type II. Genomics 1997; 42: 38-45. 31. Crosby AH, Edwards SJ, Murray JC, Dixon MJ: Genomic organization of the human osteopontin gene: exclusion of the locus from a causative role in the pathogenesis of dentinogenesis imperfecta type II. Genomics 1995; 27:155-160. 32. Crosby AH, Scherpbier-Heddema T, Wijmenga C et al.: Genetic mapping of the dentinogenesis imperfecta type II locus. Am JHum Genet 1995; 57: 832-839. 33. Zhang X, Chen L, Liu J et al.: A novel DSPP mutation is associated with type II dentinogenesis Imperfecta in a chinese family. BMC Medical Genetics 2007; 8: 52. 34. Suzuki S, Sreenath T, Haruyama N et al.: Kulk Dentin sialoprotein and dentin phosphoprotein have distinct roles in dentin mineralization. Matrix Biol 2009; 28 (4): 221-229. 35. Hursey RJ Jr., Witkop CJ Jr., Miklashek D, Sackett LM: Dentinogenesis imperfecta in a racial isolate with multiple hereditary defects. Oral Surg 1956; 9: 641-658. 36. Bai H, Agula H, Wu Q et al.: A novel DSPP mutation causes dentinogenesis mperfecta type II in a large Mongolian family Bai et al. BMC Medical Genetics 2010; 11: 23. 37. Cole D, Cohen MM: Osteogenesis imperfecta: an update. J Pediatr 1991; 115: 73-74. 38. Gage JP, Symons A, Roumaniuk K, Daley T: Hereditary opalescent dentine: variation in expression. J Dent Child 1991; 58: 134-139. 39. O’Carroll MK, Duncan W, Perkins T: Dentin dysplasia: review of the literature and a proposed sub classification based on radiographic findings. Oral Surg, Oral Med, Oral Pathol 1991; 72: 119-125. 40. Seow WK: Clinical diagnosis and management strateges of amelogenesis imperfecta variants. Pediatr Dent 1993; 15: 384-393.
