© Borgis - Postępy Nauk Medycznych 9/2014, s. 652-657
*Anna Bochyńska1, Lidia Ziółkowska2
Kardiomiopatia przerostowa u noworodka z rzadkim zespołem genetycznym – trudności diagnostyczne i terapeutyczne
Hypertrophic cardiomyopathy in a newborn with a rare genetic syndrome – diagnostic and therapeutic difficulties
1Dział Diagnostyki Kardiologicznej, Wojewódzki Szpital Zespolony im. L. Rydygiera w Toruniu – Szpital Dziecięcy, Toruń
Kierownik Oddziału: lek. med. Grażyna Zadrozińska
2Klinika Kardiologii, Instytut „Pomnik – Centrum Zdrowia Dziecka”, Warszawa
Kierownik Kliniki: prof. dr hab. med. Wanda Kawalec
Streszczenie
Kardiomiopatia przerostowa charakteryzuje się przerostem mięśnia lewej komory serca, przy braku innej patologii, takiej jak nadciśnienie tętnicze lub wrodzona wada serca. Objawy kliniczne zazwyczaj zależą od wieku dziecka w momencie rozpoznania choroby. U noworodków i niemowląt najczęściej występują objawy niewydolności serca i słaby przyrost masy ciała. Początek objawów w okresie niemowlęcym jest czynnikiem źle rokującym, a postępujący przerost mięśnia sercowego oraz występowanie złożonych komorowych i nadkomorowych zaburzeń rytmu serca stanowi istotny problem terapeutyczny. Zespół Leopard jest rzadkim zespołem genetycznym, dziedziczącym się autosomalnie dominująco. W większości przypadków jego przyczyną jest mutacja w niereceptorowym genie PTPN11. Charakteryzuje się szerokim spektrum objawów klinicznych i anomalii rozwojowych. U około 85% pacjentów z zespołem Leopard występuje patologia serca, w tym u około 20% dzieci to kardiomiopatia przerostowa. Dotychczas w literaturze opisano około 200 pacjentów z zespołem Leopard. Przedstawiamy przypadek 8-miesięcznego niemowlęcia, u którego wystąpiła wczesna manifestacja kliniczna (po raz pierwszy w okresie noworodkowym) rzadkiego zespołu chorobowego powstałego w wyniku mutacji genu PTPN11, z dominującymi objawami pogłębiających się zaburzeń czynności układu krążenia z powodu kardiomiopatii przerostowej.
Summary
Hypertrophic cardiomyopathy is characterized by left ventricular hypertrophy with the absence of other cardiac anomaly as arterial hypertension or congenital heart disease. Clinical symptoms usually depend on the age of a child when the disease is recognized. The newborns and infants most often develop heart failure and weak weight gain. Diagnosis and clinical manifestation of the disease in infancy is associated with poor prognosis. Progressive hypertrophy and occurrence of the ventricular and supraventricular arrhythmias are major therapeutic problems. Leopard syndrome is a rare autosomal dominant genetic disorder. The majority of cases is caused by mutation in non-receptor gene PTPN11. It is characterized by a broad spectrum of clinical symptoms and organ malformations. In approximately 85% patients with Leopard syndrome the cardiac pathology occurs, including in about 20% of cases hypertrophic cardiomyopathy. So far in the literature about 200 patients with Leopard syndrome have been reported. We present a case of an 8-month old infant in whom were early clinical manifestation (the first time in the neonatal period) a rare syndrome resulting from mutations of the gene PTPN11, with predominant symptoms deepening cardiovascular dysfunction due to hypertrophic cardiomyopathy.

Wstęp
Kardiomiopatia przerostowa (ang. Hypertrophic cardiomyopathy – HCM) jest chorobą charakteryzującą się przerostem mięśnia lewej komory, który nie jest wtórny do wrodzonej wady serca lub nadciśnienia tętniczego (1). Występuje z częstością około 0,2% populacji (1/500 rocznie), porównywalnie u płci męskiej i żeńskiej (1, 2). Kardiomiopatia przerostowa jest chorobą dziedziczoną w sposób autosomalny dominujący lub może powstawać jako mutacja „de novo” (1-3). HCM może występować u pacjentów z zespołami klinicznymi, takimi jak wrodzone błędy metabolizmu, lizosomalne choroby spichrzeniowe – choroba Pompe, choroba Danona, choroba Fabry oraz z zespołami genetycznymi – zespół Noonan, Leopard, zespół Beckwitha-Wiedemanna oraz chorobami nerwowo-mięśniowymi (ataxia Friedricha) (4). HCM jest chorobą o bardzo różnorodnym przebiegu klinicznym, dlatego też zarówno wyniki leczenia, jak i rokowanie są trudne do przewidzenia (5). Przerost mięśnia sercowego może pojawić się zaraz po urodzeniu lub w pierwszych latach życia, lecz najczęściej ujawnia się w okresie dojrzewania i postępuje wraz ze wzrostem somatycznym dziecka, co różni się w porównaniu z pacjentami dorosłymi (5). Według danych z piśmiennictwa, rozpoznanie kardiomiopatii przerostowej w wieku niemowlęcym postawiono u 10 do 36% pacjentów (5), natomiast w okresie noworodkowym choroba rozpoznawana jest znacznie rzadziej. Objawy kliniczne choroby zazwyczaj zależą od wieku dziecka w momencie rozpoznania HCM. U noworodków i niemowląt występują zazwyczaj objawy niewydolności serca i słaby przyrost masy ciała. Początek objawów w okresie niemowlęcym jest czynnikiem źle rokującym, a postępujący przerost mięśnia lewej oraz często również prawej komory, występowanie złożonych komorowych i nadkomorowych zaburzeń rytmu serca stanowi istotny problem terapeutyczny (1, 2, 6). Kardiomiopatia przerostowa jest drugą co do częstości występowania (po zwężeniu zastawki tętnicy płucnej) patologią serca w zespole Noonan lub w jego allelicznej postaci – zespole Leopard (7). Dotychczas w literaturze opisano około 200 przypadków zespołu Leopard (8). W dostępnym piśmiennictwie polskim nie znaleziono opisu przypadku współistnienia kardiomiopatii przerostowej i zespołu Leopard rozpoznanego już w wieku noworodkowym.
Opis przypadku
Aktualnie 8-miesięczne niemowlę płci żeńskiej, z CI, PI, (ciąża powikłana w 24 tygodniu infekcją dróg moczowych u matki), urodzone w 37 tygodniu ciąży siłami natury, ocenione na 8-9-9-9 punktów w skali Apgar, masa urodzeniowa 3250 gramów. U matki stwierdzono nosicielstwo paciorkowca grupy B (GBS+) – nie wdrożono pełnej profilaktyki przed porodem ze względu na odpłynięcie wód płodowych w domu. Dziecko skierowano do Szpitala Dziecięcego im. L. Rydygiera w Toruniu w 3. dobie życia z powodu wysłuchanego szmeru nad sercem oraz sinicy obwodowej (SAT 97%). W badaniu przedmiotowym zwracały uwagę dyskretne cechy dysmorfii twarzy – hiperteloryzm, nisko osadzone małżowiny uszne, szeroka płaska nasada nosa. Nad całą okolicą przedsercową słyszalny był szmer skurczowy o głośności 3/6 w skali Levine’a. Wątroba była wyczuwalna około 1,5 cm poniżej prawego łuku żebrowego. Ze względu na obserwowaną męczliwość oraz duszność dziecka podczas karmienia piersią zastosowano karmienie z butelki. W badaniach laboratoryjnych z odchyleń od normy stwierdzono znacznie podwyższony poziom NT-pro BNP > 35 000 pg/ml (norma < 125 pg/ml), stopniowo narastające wartości troponiny I (0,079 ug/l-0,187 ug/l; norma < 0,04 ug/l) oraz nieznacznie podwyższone stężenie frakcji sercowej kinazy kreatyny (6,03 ng/ml; norma < 5 ng/ml). W pozostałych badaniach stwierdzono podwyższone stężenie amoniaku 188,9 ug/dl (norma 42-179 ug/dl) oraz hiperbilirubinemię. W badaniu elektrokardiograficznym stwierdzono rytm zatokowy, miarowy 120/min, cechy przerostu prawej i lewej komory, blok przedsionkowo-komorowy I stopnia (PQ = 0,14 sek). W badaniu radiologicznym klatki piersiowej opisano powiększoną sylwetkę serca, wskaźnik sercowo-płucny 0,65 oraz nieco wzmożony rysunek naczyniowy w górnych partiach płuc.
W badaniu echokardiograficznym serca wykonanym w 3. i 6. dobie życia stwierdzono masywny przerost mięśniówki lewej oraz prawej komory – grubość ściany tylnej lewej komory wynosiła 177% średniej normy w stosunku do BSA (6,4 mm; norma 2,7-4,9 mm), grubość przegrody międzykomorowej 204% średniej normy w stosunku do BSA (8,6 mm; norma do 6,1 mm). Przypodstawna część przegrody międzykomorowej wpuklała się w drogę odpływu lewej komory oraz stwierdzono występowanie zjawiska SAM przedniego płatka zastawki mitralnej (SAM = śródskurczowe przemieszczanie się i przyleganie przedniego płatka zastawki mitralnej do przerośniętej przegrody międzykomorowej). Na podstawie badania echokardiograficznego rozpoznano kardiomiopatię przerostową (ryc. 1).
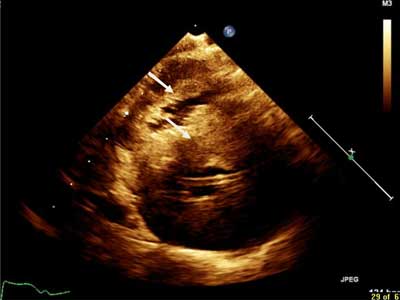
Ryc. 1. Przerost przegrody międzykomorowej i mięśnia prawej komory serca (strzałki). Projekcja echokardiograficzna przymostkowa w osi krótkiej na poziomie zastawki mitralnej.
W kilkakrotnie wykonywanych 24-godzinnych zapisach EKG metodą Holtera stwierdzono prawidłową średnią czynność serca we śnie oraz w czuwaniu, pojedyncze pobudzenia dodatkowe nadkomorowe, okresowo blok p-k I stopnia (PQ = 0,14-0,16 s), QTc było prawidłowe (0,35-0,41 s). W celu ustalenia etiologii HCM wykluczono obecność patogennych mutacji w obrębie genu łańcucha ciężkiego beta-miozyny. Wyniki badań w kierunku chorób metabolicznych – profil aminokwasów i acylokarnityn w suchej kropli krwi metodą tandemowej spektroskopii mas (MS/MS) oraz profil kwasów organicznych w moczu metodą GC/MS były prawidłowe. Wykluczono również obecność choroby Pompe. W badaniu kariotypu dziecka nie stwierdzono nieprawidłowości. Wywiad rodzinny nie był obciążony, nie stwierdzono występowania HCM u rodziców ani u innych członków rodziny. W badaniu okulistycznym nie stwierdzono obecności charakterystycznych dla nerwiakowłókniakowatości typu 1 guzków Lischa w obrębie tęczówek. Z uwagi na cechy dysmorfii twarzy oraz pojawiające się plamy soczewicowate na tułowiu wysunięto podejrzenie zespołu Noonan lub jego allelicznej postaci zespołu Leopard. W badaniu genetycznym wykonanym w 6. miesiącu życia stwierdzono obecność patogennej mutacji p.Gln510Glu (c.1528C > G) w jednym allelu genu PTPN11, co potwierdziło kliniczne rozpoznanie zespołu Noonan lub jego allelicznej postaci zespołu Leopard. W trakcie kolejnych miesięcy dziewczynka była kilkakrotnie hospitalizowana z powodu stopniowo narastających objawów niewydolności serca oraz stwierdzanych w badaniu EKG metodą Holtera epizodów częstoskurczów nadkomorowych o częstości 160-200/min (ryc. 2).
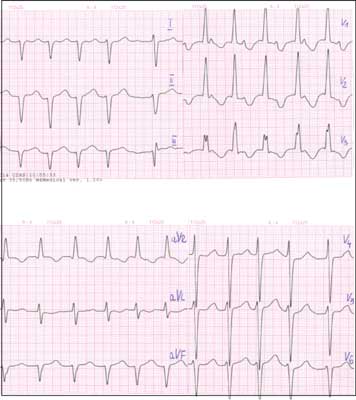
Ryc. 2. Częstoskurcz nadkomorowy 160/min u pacjentki z HCM i zespołem Leopard (EKG przesuw papieru 50 mm/s).
Powyżej zamieściliśmy fragment artykułu, do którego możesz uzyskać pełny dostęp.
Mam kod dostępu
- Aby uzyskać płatny dostęp do pełnej treści powyższego artykułu albo wszystkich artykułów (w zależności od wybranej opcji), należy wprowadzić kod.
- Wprowadzając kod, akceptują Państwo treść Regulaminu oraz potwierdzają zapoznanie się z nim.
- Aby kupić kod proszę skorzystać z jednej z poniższych opcji.
Opcja #1
29 zł
Wybieram
- dostęp do tego artykułu
- dostęp na 7 dni
uzyskany kod musi być wprowadzony na stronie artykułu, do którego został wykupiony
Opcja #2
69 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 30 dni
- najpopularniejsza opcja
Opcja #3
129 zł
Wybieram
- dostęp do tego i pozostałych ponad 7000 artykułów
- dostęp na 90 dni
- oszczędzasz 78 zł
Piśmiennictwo
1. Kawalec W, Ziółkowska L, Turska-Kmieć A: Kardiomiopatie. [W:] Kubicka K, Kawalec W: Kardiologia dziecięca. Tom 2, PZWL 2003; 17: 789-797.
2. Ziółkowska L, Kawalec W, Turska-Kmieć A et al.: Postępy w diagnostyce i leczeniu kardiomiopatii przerostowej u dzieci. Stand Med 2006; 3: 147-151.
3. Ziółkowska L, Kawalec W, Pręgowska K et al.: Zespół genetyczny u pacjentki z kardiomiopatią przerostową i przetrwałym przewodem tętniczym. Stand Med 2005; T7, 23: 87-90.
4. Shiraz A: Maskatia. Hypertrophic Cardiomyopathy: Infants, Children, and Adolescents. Congenital Heart Disease 2012; 7: 84-92.
5. Ziółkowska L, Turska-Kmieć A, Boruc A et al.: Obraz kliniczny kardiomiopatii przerostowej u dzieci – doświadczenia własne. Post Nauk Med 2011; 12: 1008-1014.
6. Gołąbek M, Wróblewska-Kałużewska M, Brzeszkiewicz K: Pierwotna kardiomiopatia przerostowa lewej komory serca jako stan zagrożenia życia. Pediatr Pol 2004; LXXIX, 4: 279-285.
7. Hoban R, Roberts AE, Demmer Let al.: Noonan syndrom due to a SHOC2 mutation presenting with fetal distress and fatal hypertrophic cardiomyopathy in a premature infant. Am J Med Genet 2012; 158A, 6: 1411-1413.
8. Sarkozy A, Digilio MC, Dallapiccola B: LEOPARD syndrome. Orphanet J Rare Dis 2008; 3: 13.
9. Digilio MC, Sarkozy A, de Zorzi A et al. Leopard syndrome: clinical diagnosis in the first year of life. Am J Med Genet A 2006; 140(7): 740-746.
10. Sarkozy A, Conti E, Digilio MC et al.: Clinical and molecular analysis of 30 patients with multiple lentigines Leopard syndrome. J Med Genet 2004; 41(5): e68.
11. Klapecki J, Obersztyn E, Łaniewski-Wołłk M et al.: Zmienność ekspresji klinicznej zespołu Noonan – analiza dwóch przypadków rodzinnych. Wiad Lek 2008; LXI, 1-3: 74-80.
12. Voron DA, Hartfield HH, Kalkhoff MD: Multiple lentigines syndrome. Case report and review of the literature. Am J Med 1976; 60: 447-456.
13. Gołąbek M, Wróblewska-Kałużewska M, Przybylski A: Nagłe zatrzymanie krążenia z następowym wszczepieniem kardiowertera-defibrylatora u dziecka z kardiomiopatią przerostową. Przegl Pediatr 2004; 34(3/4): 250-253.
14. Ziółkowska L, Kawalec W, Turska-Kmieć A et al.: Czynniki ryzyka nagłego zgonu sercowego u dzieci z kardiomiopatią przerostową. Stand Med/Pediatr 2009; 6: 823-828.
15. Wilkinson JD, Lowe AM, Salbert BA et al.: Outcomes in children with Noonan syndrome and hypertrophic cardiomyopathy: a study from the Pediatric Cardiomyopathy Registry. Am Heart J 2012; 164, 3: 442-448.
