© Borgis - New Medicine 2/2002, s. 80-82
P. Beighton1, K. Kozlowski2
A new, distinctive from of spondylo-epi-metaphyseal dysplasia
1Department of Human Genetics, Cape Town, S. Africa
2Department of Medical Imaging, New Children´s Hospital, Sydney, Australia
Summary
Spondylo-epi-metaphyseal dysplasias are a group of complex disorders, most of which defy classification. We report a patient with distinctive radiographic features who presents a previously undescribed form of spondylo-epi-metaphyseal dysplasia.
INTRODUCTION
The spondylo-epi-metaphyseal dysplasias (SEMD) comprise an extensive group of different disorders which share the common characteristics of spinal, metaphyseal and epiphyseal involvement (1). A few SEMDs, such as metatropic dysplasia, diastrophic dysplasia or pseudoachondroplasia have well-recognised phenotypic and radiographic diagnostic features. The majority of affected patients, however, defy classification because of the rarity of these conditions, non-specific phenotypes, poor radiographic documentation, normal biochemical investigations and uncharacteristic histology. Detailed radiographic description is the first step in the classification of this complex group of conditions. In order to exemplify this situation, we report a boy with an unclassifiable SEMD and describe our approach to differential diagnosis.
CASE REPORT
MA, a male was born to young, healthy, unrelated parents after an uneventful pregnancy and breech delivery. At birth, he was noted to have short limbs, CTEV and a hydrocele. The parents and two elder siblings were healthy and had normal stature; the family history was unremarkable.
When examined at the age of 3 years ha was found to be a healthy child with a normal intellect. Short stature with proximal shortening of the limbs and marked lumbar hyperlordosis were evident. His elbows lacked 10 degrees of full extension and he wore surgical boots and calipers for a residual CTEV. Apart from 5th finger clinodactyly, his hands were normal. The face was normal there were no systemic ramifications. Routine laboratory investigations including cytogenetic studies and urinary screening for mucopolysaccharides yielded normal results.
Radiographic at the age of 3 1/2 months revealed distinctive skeletal changes, notably delayed bone age and metaphyses of the tubular bones which were cupped, with pointed edges. The distal ends of the femora and the acetabulae had a trident configuration (Fig 1A-C). A lateral view of the spine obtained at the age of 15 months showed hypoplasia of the 12th thoracic vertebra (Th12), with some posterior displacement of the vertebral body. The other vertebral bodies were slightly oval in shape (Fig 2). The skull was normal.
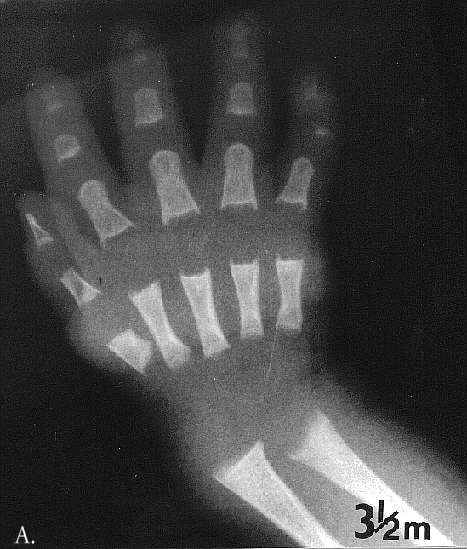
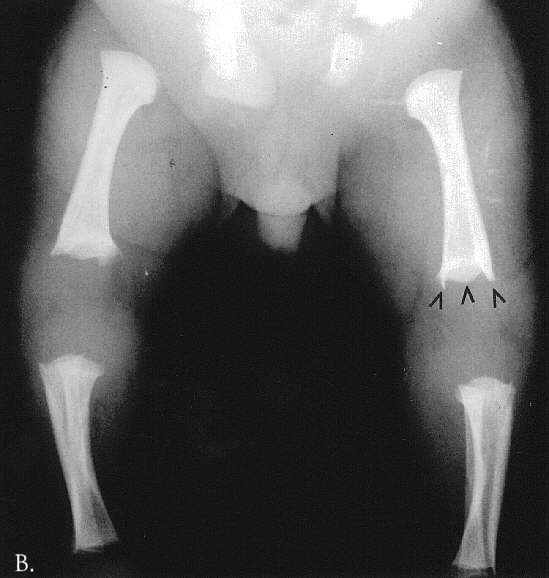
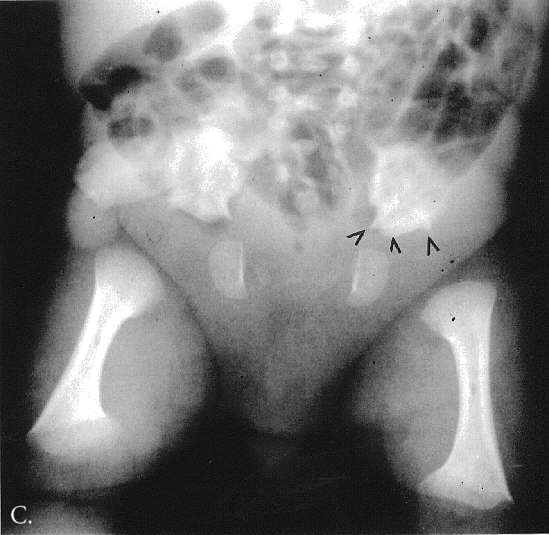
Fig. 1A-C. MA at the age of 3,5 months. A. Hand. Cupped metaphyses with pointed edges, shortened 1st, 2nd and 5th fingers. Hypoplastic/dysplastic middle and distal phalanges and 1st metacarpal. Distal shortening of the ulna. B. Lower extremities. Delayed bone age – "empty” knees. Trident configuration of the distal femora (arrowed). C. Pelvis. Club-shaped proximal femoral metaphyses. Tri-radiate acetabulum (arrowed). Hypoplastic pubic bones.
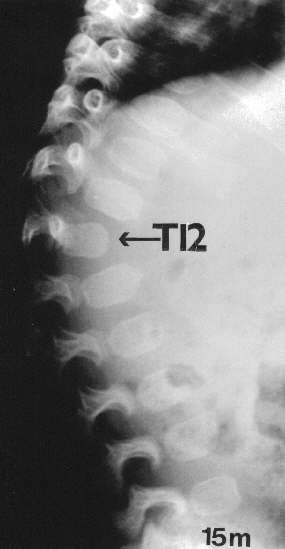
Fig. 2. MA at the age of 15 months. Kyphosis, hypoplasia of Th12 and oval vertebral bodies.
By the age of 3,5 years, all the epiphyseal ossification centres were slightly hypoplastic/dysplastic and the carpal bone age was still delayed. Otherwise, bone age had reverted to normality. The 1st, 2nd and 5th digits were shoetened and a Kirner anomaly was present in the middle phalanx of the little finger. The growth cartilage of the tubular bones of the hands was widened and there was distal shortening of the ulna. A distinctive feature in the feet was the presence of calcaneal spurs. In the pelvis the acetabulum and the growth cartilages were horizontal, the femoral necks were short and the capital femoral epiphyses were small and flattened. The trident shape of the acetabulum was no longer present, the bodies of the iliac bones were short and the ilial wings were rounded. The anerior ends of the ribs were widened and the scapulae were wide with prominence at their lower edges (Fig 3A-E).
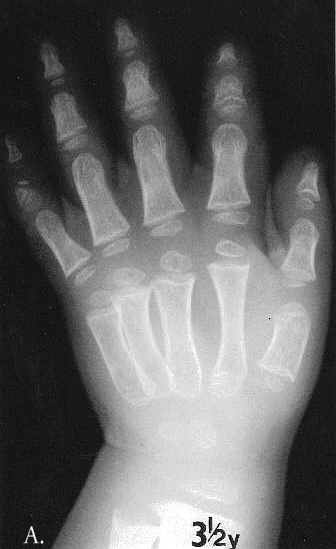
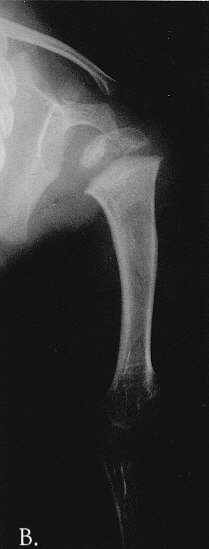
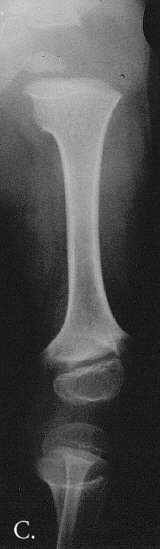
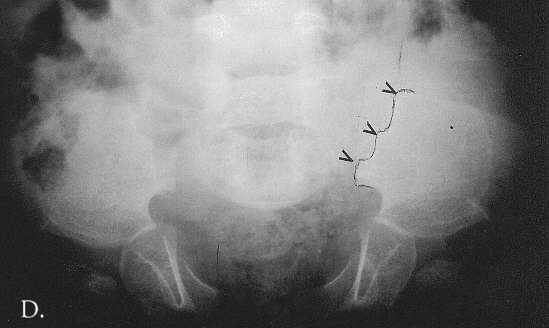
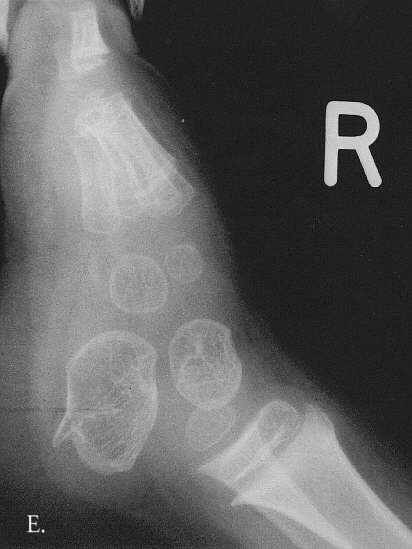
Fig. 3A-E. A. Hand and Forearm. Cupped metaphyses and flattened epiphyses in the tubular bones. B. Left humerus. Moderately severe meta-epiphyseal changes at the proximal end of the humerus. C. Left femur and hip joint. Horizontal configuration of the acetabulum and proximal region of the wide, short femoral neck. The capital femoral epiphyses is flattened and positioned medially in relation to the femoral neck. The fibula is elongated proximally. D. Pelvis. The outer edge of the iliac wing is rounded with a slightly irregular outline. The medial edge is oblique and has a trident apperance (arrowed). The iliac body is short and wide. E. Foot. A large spur is evident at the posterior, lower aspect of the calcaneus.
DISCUSSION
Our patient has an hitherto unreported SEMD and review of the literature did not reveal any similar cases (2, 3). The distinctive features in our patient are:
1. Markedly delayed neonatal bone age which normalises with age.
2. Trident acetabulum and trident configuration of the distal femora. At the age of 3 1/2 years the iliac wings were prominent with slightly irregular lateral and superior medial borders. The iliac bodies were short. The acetabulae and proximal ends of the femora were horizontal and coxa valga was present.
3. The twelfth thoracic vertebral body was hypoplastic and posteriorly displaced. The other vertebral bodies were oval and large calcaneal spurs were present.
The differential diagnosis of the condition in our patient includes disorders presenting in the neonatal/infantile period with markedly delayed bone age, disorders with a trident acetabulum and disorders with kyphosis secondary to hypoplastic lumbar/thoracic vertebral bodies.
Severely retarded bone age in neonates and infants occurs in hypothyroidism, neonatal lethal bone dysplasias, spondylometaphyseal and spondylepimetaphyseal dysplasias. Hypothyroidism can easily be excluded as cupping of the metaphyses and trident acetabulum are not features of this condition. Moreover, hypoplasia of the vertebral bodies affects the upper lumbar spine and not Th12 and the shapes of the vertebrae are different from those of our patient. Finally, normal mental development is not a feature of severe hypothyroidism (2).
Trident acetabulum is a distinctive feature of several bone dysplasias such as achondroplasia, chondroectodermal dysplasia, asphyxiating thoracic dystrophy and thanatophoric dysplasia. Each of them, however, is a distinct entity with clinical and radiographic features which are so characteristic that it is difficult to confuse them with other disorders (1).
Vertebral kyphosis secondary to hypoplasia/dysplasia of one or two vertebral bodies is a common feature of some disorders of complex carbohydrate metabolism (mucopolysaccharidoses, mucolipidoses). In these conditions the upper lumbar vertebral bodies and the rest of the skeleton shows changes descriptivelly termed "dysostosis multiplex”; these are quite different from the skeletal abnormalities in our patient.
In the SEMDs, the epiphyses, metaphyses and the spine are affected. There is similar involvement in the patient whom we described but the pattern of his changes is unique. The characteristic features in this boy are the trident acetabulum and the trident shape of the distal femoral metaphyses which is present at birth but disappears with age. The cupped metaphyses with pointed edges at birth are another distinctive feature. The appearances of the bony structures around the hip joints changes during the first few years of life; in particular the acetabulae, the proximal femoral metaphyses and the adjacent growth cartilages become horizontal and the flattened capital femoral epiphyses are positioned in the medial portion of the short femoral necks. The medial edge of the iliac bone has a trident configuration. Hypoplasia of the 12th vertebral body and the large spurs on the plantar aspect of the calcaneal bones are additional unusual features.
The Irapa and the Iraqi types of SEMD superficially resemble the condition present in our patient. They differ in that the Irapa type shows generalised platyspondyly and coxa vara – features which are absent in our patient (4), while the Iraqi type resembles at early age achondroplasia (5). Neither of them have hypoplasia//dysplasia of the Th12 vertebral body and calcaneal spurs (3).
CONCLUSION
We have documented a South African boy with a form of SEMD with distinctive radiographic appearances. Diagnosis in this group of conditions is dependent upon the pattern and evolution of skeletal abnormalities, and it is appropriate for periodical full skeletal survey to be performed in persons with SEMD. Detailed description of the radiographic changes is the first step in the classification of this complex group of bone dysplasias.
Acknowledgements
We are grateful for support from the South African Orthopaedic Association.
Piśmiennictwo
1. Kozlowski K., Beighton P.: Gamut Index of Skeletal Dysplasias. 3rg Ed 2001, Springer Verlag, LondonBerlin. 2. Maroteaux P.: Les maladies osseuses de l´enfant. 3-e Ed 1995, Flammarion Paris. 3. Taybi H., Lachman R.S.: Radiology of syndromes, metabolic disorders, and skeletal dysplasias. 4th Ed 1996, Mosby St Louis. 4. Hernandez Z. et al.: Autosomal recessive spondylo-epi-metaphyseal dysplasia (Irapa type) in a mexican family: Delineation of the syndrome. Am. J. Med. Genet. 1980, 5:179-188. 5. Figuera L.E. et al.: Spondyloepimetaphyseal dysplasia (SEMD). Shohat Type. Am. J. Med. Genet. 1994, 51:213-215.
